引言
去甲腎上腺素 (Norepinephrine 或 noradrenaline; NE ) 、 多巴胺 (Dopamine; DA ) 和 血清素 (Serotonin ;或 5-羥色胺 5-hydroxytryptamine, 5-HT ) 是重要的單胺類神經遞質,影響著包括運動、激素分泌、獎懲和情緒相關的復雜行為和生理過程。這些神經遞質在突觸前膜 去極化 後釋放到突觸間隙,隨後啟用突觸後膜上的受體並觸發影響神經元興奮性、突觸可塑性和各種生理過程的細胞內訊號級聯。正常生理情況下,從突觸前神經元釋放的神經遞質,在完成訊號傳導後必須及時高效地從突觸間隙被清除,從而實作訊號終止。透過突觸前神經元或者神經膠質細胞質膜上的 單胺類神經遞質轉運蛋白 (Monoamine transporters; MATs ) ,可以將單胺類神經遞質再攝取回突觸前膜從而終止其對下遊受體的啟用狀態。目前,MATs家族抑制劑主要在醫藥市場上用作抗抑郁藥,可根據其化學結構和作用方式分為不同的類別 (圖1) 。
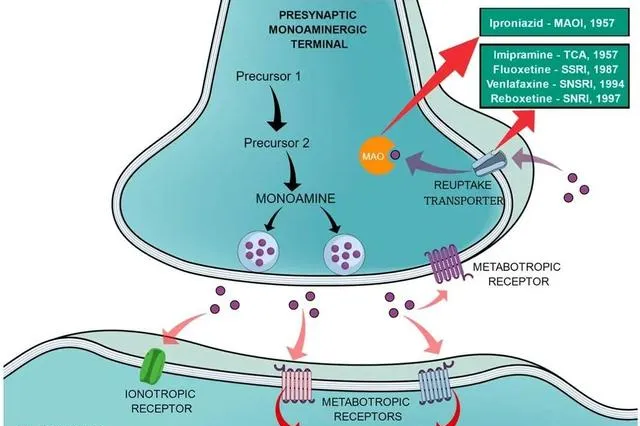
圖 1 單胺能神經元中神經遞質的傳遞過程及不同抗抑郁藥物的作用方式(Credit: Acta Neuropsychiatr ) 【1】
MATs 屬於 神經遞質鈉同向轉運蛋白 (Neurotransmitter: sodium symporter; NSS ) 家族成員,其利用 膜外/內鈉離子Na + 的濃度梯度 來完成對神經遞質的同向跨膜運輸,這一過程可能依賴於 氯離子Cl - 同向轉運以及鉀離子K + 的反向轉運 。近年來對MATs 中受質轉運及抗抑郁藥物調節機制的研究進展迅速,其中果蠅多巴胺轉運蛋白dDAT和人源血清素轉運蛋白SERT的不同構象以及與多種抗抑郁藥物分子結合的結構已經得到解析。但關於人源去甲腎上腺素轉運蛋白NET的結構研究相對進展緩慢,這大大阻礙了抗抑郁藥物的進一步最佳化和開發。
2024年7月24日,清華大學生命科學學院/北京生物結構前沿研究中心 閆創業/袁亞飛 團隊在 Nature 雜誌發表了題目為 Molecular basis of human noradrenaline transporter reuptake and inhibition」 (人類去甲腎上腺素轉運蛋白再攝取與抑制分子基礎) 的研究論文。該工作透過結構生物學和生物化學方法 闡明了NET轉運受質NE和DA的機制,首次報道了NET中的第二個受質結合位點和NSS家族的鉀離子結合位點,揭示了四種不同類別的常用上市抗抑郁藥物的選擇性抑制機制,為進一步開發靶向MATs的藥物奠定了基礎。

該團隊利用冷凍電鏡技術成功解析了人源NET蛋白的八個高分辨率結構,包括天然狀態、結合受質( NE 、 DA )和四種常用上市抗抑郁藥物 ( 托莫西汀 :Atomoxetine,ATX; 地昔帕明 :Desipramine,DSP; 艾司西酞普蘭 :Escitalopram,ESC; 安非他酮 :Bupropion,BPP) 的結合狀態。這些結構是天然蛋白在未使用任何基準標記或突變的情況下獲得,分辨率在2.5到3.5埃之間 (圖2) 。
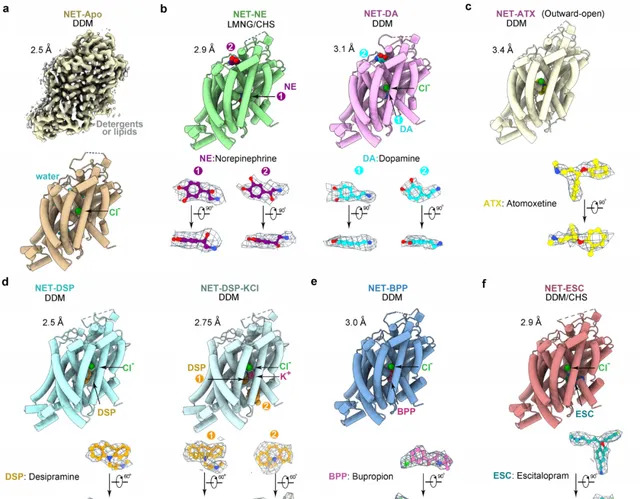
圖2. 人源NET天然狀態、結合兩種受質和四種抗抑郁藥物復合物的冷凍電鏡結構(Credit: Nature )
研究發現 NET中央口袋S1處辨識NE或DA的方式與報道的果蠅dDAT不同,其 兒茶酚環上的兩個羥基主要透過水介導的氫鍵辨識 (圖3a,b) 。此外,研究在 NET與兩種受質結合的結構中都觀測到了第二個受質的密度 (S2位點) 。值得註意的是,與已報道的SERT中的S2位點顯著不同,NET中的S2位點位於TM1b C末端,且僅存在於向內閉合或開放的構象中,向外開放構象轉變會導致S2位點被破壞 (圖3c) 。透過生化實驗發現,S2位點內的某些突變顯著影響轉運蛋白的轉運速度。推測NET中S2位點的功能可能是提前載入受質,從而加快受質轉運效率。
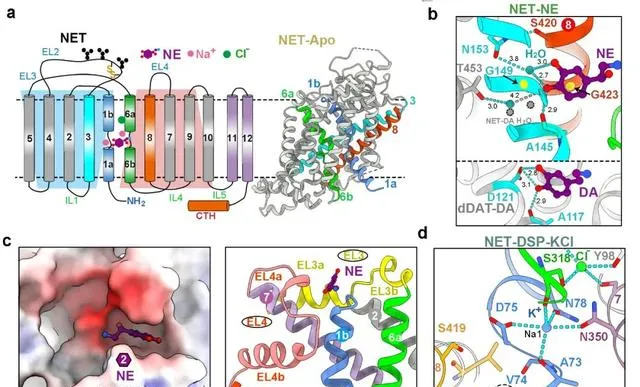
圖3. NET中心口袋S1的受質結合方式( a-b )、S2位點( c )及K + 離子結合位點( d )(Credit: Nature )
MATs成員是離子耦合的膜蛋白,作為次級主動轉運蛋白,它們共享兩個保守的Na + 結合位點 (Na1和Na2) 和一個保守的Cl - 結合位點,利用跨膜離子梯度的能量來催動神經遞質的運輸 (圖3a) 。早在1979年時K + 離子就被提出參與MATs的轉運迴圈,但直到現在實驗結構仍未確定特定的K + 結合位點 【3】 。研究人員在向內開口的NET-DSP-KCl結構的 Na1 位點處發現了K + 的額外密度,而該密度在 NaCl 條件下的NET內向開口構象中並不存在 。據此,該研究 首次透過結構驗證了NSS 家族中 K + 離子的結合位點。 這一K + 離子位點恰好是Na1位點,該位點在向內開口釋放Na + 離子之後,局部殘基構象發生改變,使得該位點更偏好螯合K + 離子 (圖3d) 。盡管確認了 Na1 處 K + 的存在及其在運輸中的作用,但其觸發構象轉變的確切機制仍需要進一步研究。
深入了解 不同抗抑郁藥物對MATs的選擇性抑制 的分子基礎對於抗抑郁藥物的開發和套用具有重要意義。 托莫西汀 是一種NE再攝取抑制劑 (NRI) ,其對 NET 的選擇性比 SERT 和 DAT 高約 30 倍和 130 倍; 地昔帕明 是三環類抗抑郁藥 (TCA) ,其對NET的選擇性比SERT高約20倍; 安非他酮 是典型的NE和DA再攝取抑制劑 (NDRI) ;而 艾司西酞普蘭 是一種典型的選擇性血清素再攝取抑制劑 (SSRI) ,其對SERT的選擇性比NET高約2600倍。該研究透過解析四種不同類別的抗抑郁藥物的高分辨率結構,為理解其對NET、DAT和SERT選擇性提供了分子基礎。透過不同抑制劑濃度的[3H]-NE攝取測定,該研究進一步揭示了托莫西汀的競爭性抑制及其它三種抑制劑的混合型抑制機制與結合不同構象相關。這些研究為理解靶向MATs的抗抑郁藥物選擇性提供了重要見解,MATs抗抑郁藥獨特的結合模式和多靶點特性值得制藥行業特別關註。
綜上,研究人員提出了 NET的轉運過程模型 (圖4) 。在向外開放的構象中,Na + 、Cl - 和受質NE與中央口袋結合,啟動 NET 的閉合,進入閉合狀態。在這個轉變過程中,第二個受質可以提前結合到新形成的膜外S2位點中。隨後,TM1a 的開啟 (向內開放構象的標誌特征) 促進受質與兩個Na + 一起從中央口袋釋放。在轉變回向外開放構象的過程中,S2 位點的局部結構將受到破壞,導致S2位點處受質的釋放。釋放的受質可能直接進入到中央口袋,開始下一個轉運迴圈。這一過程與兩個 Na + 離子和一個 Cl - 離子進入細胞質以及可能一個 K + 離子進入細胞外基質的轉運耦合。而不同類別的抗抑郁藥物占據 S1 位點阻礙受質轉運。
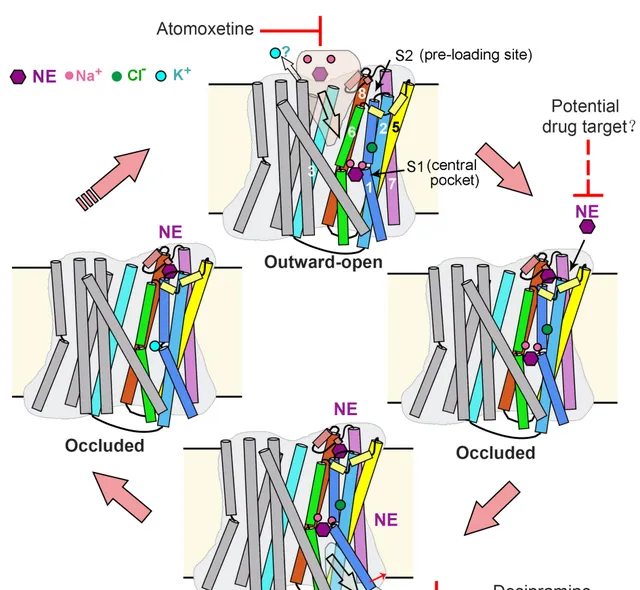
圖4. NET的交替開放轉運模型和抑制機制(Credit: Nature )
該研究亮點 : 首次發現了NET的第二個受質結合位點以及NSS家族的K + 結合位點,揭示了NET受質辨識機制、交替開放轉運模型、以及不同類別抗抑郁藥物選擇性抑制的分子機制,為抗抑郁藥物的進一步研發提供了基礎。
參考文獻
1.Pereira, V. S. & Hiroaki-Sato, V. A. A brief history of antidepressant drug development: from tricyclics to beyond ketamine. Acta Neuropsychiatr 30, 307-322 (2018).
2. Cheng, M. H. & Bahar, I. Monoamine transporters: structure, intrinsic dynamics and allosteric regulation. Nat Struct Mol Biol 26, 545–556 (2019).
3.Rudnick, G. & Sandtner, W. Serotonin transport in the 21st century. J. Gen. Physiol 151, 1248-1264 (2019).
https://www.nature.com/articles/s41586-024-07719-z
責編 |探索君
排版|探索君
文章來源|「BioArt」
End











