
藥用植物因其潛在的抗腫瘤、抗炎和抗氧化特性,在民族醫學和傳統醫學中引起了極大的關註。基因組測序和合成生物學的最新進展重新激發了人們對這些天然產物的興趣。盡管有很多藥用植物的基因組和轉錄組測序數據,但缺乏可公開存取的基因註釋和表格格式的基因表現數據,這不利於它們的有效利用。為了解決這一緊迫問題,我們開發了IMP (Integrated Medicinal Plantomics)整合藥用植物組學平台(https://www.bic.ac.cn/IMP 點選閱讀原文 直接跳轉)。
IMP收錄了1007 個高品質的基因組(預期收錄所有植物的基因組,目前已收錄1007 個),整理了848,565,672 個基因,以及2,158 個轉錄組測序樣本,涵蓋了多個器官、組織、發育階段和脅迫刺激。透過整合的10 個分析模組,使用者可以簡單地在IMP中探索基因的註釋、序列、功能、分布和表達。IMP的開發和使用將會從基礎數據層面促進藥用植物分子代謝途徑的解析,進而在推動合成生物學的發展、促進藥物發現和藥物生產的天然來源的探索方面發揮重要作用。
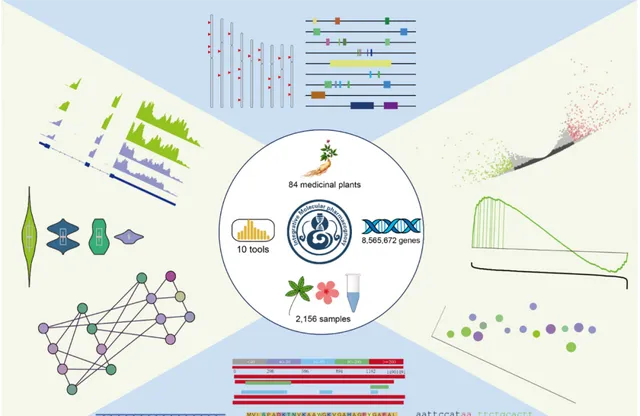
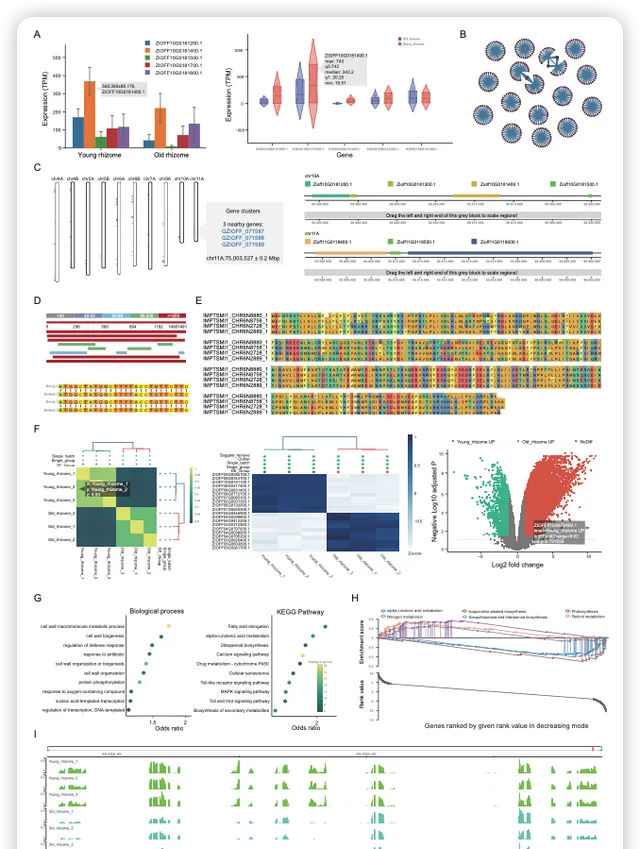
IMP 針對收錄的數據提供了 10 個功能分析模組,範例性結果如下圖,包括多基因表現圖譜的繪制、共表達基因的搜尋和鑒定、基因簇的展示、BLAST 序列搜尋、多序列比對、線上差異基因分析(樣品相關性熱圖、差異基因熱圖和火山圖)、GO/KEGG富集分析、GSEA 富集分析、IGV 基因組瀏覽器展示、引物設計、序列提取等。

具體見 NAR | 中醫科學院陳同等開發整合藥用植物組學平台 IMP
IMP 資料庫基本介紹
數據平台存取地址https://www.bic.ac.cn/IMP/。首頁采用平面組合布局,分為導航、網站描述、統計資訊和功能展示 4 個部份。
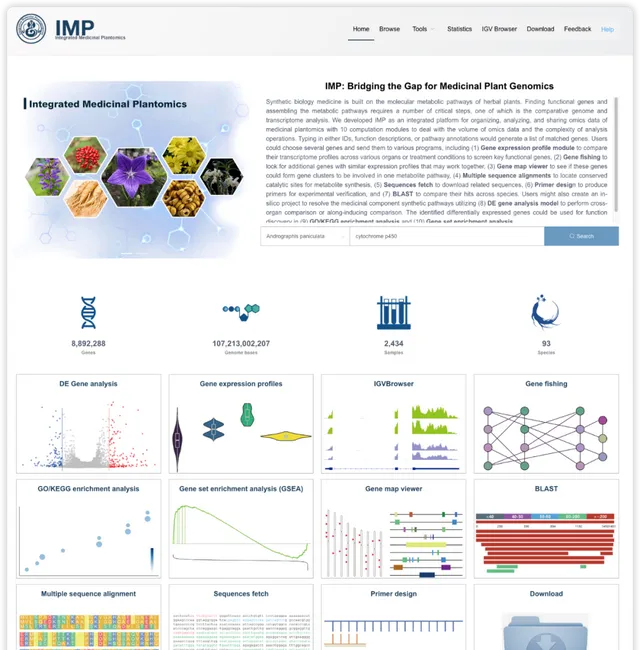
基於功能描述、註釋或基因名字的全域搜尋
在首頁的全域搜尋框中輸入基因的名字、基因的功能描述或基因的 GO 註釋/KEGG通路註釋的資訊,即輸入 任何文字 都可以去匹配出關註的基因(當然也有一些文字什麽都匹配不出來)。比如預設選中的物種是穿心蓮 ,預設輸入的文字是cytochrome p450 ,我們需要做的就是點選Submit 送出一下,新分頁會出現搜尋結果。
如果碰到頁面不出來的情況下,請看下瀏覽器最上部選單欄下面是否有 視窗被攔截 的提示。
搜尋結果頁面的標識條,會用 紅字 標記搜尋的文字資訊, 藍字 標記選擇的物種資訊。下面的表格列出所有的搜尋結果,分頁展示:
- 可以選擇一頁展示的條目數增減搜尋結果的數目,也可以選擇展示所有條目。
- 可以在右上角搜尋框進行 二次檢索 ,進一步聚焦要關註的基因。
- 右上角也可以調節表格中展示哪些列,預設只有 2 列資訊,可加列。
獨特的 Send to 快捷操作
很多物種的基因名字都是 ID 類似的編號,通常記不住。IMP 可以透過文字或序列的方式搜尋出一系列相關基因,選擇後,點選 Send to 就可以把這些目標基因集發送到對應的功能模組,實作免輸入 Gene ID的快速操作。比如檢視搜尋出的 CYP450 的整體表達資訊、基因組的分布資訊、批次序列提取、引物設計和多序列比對等。
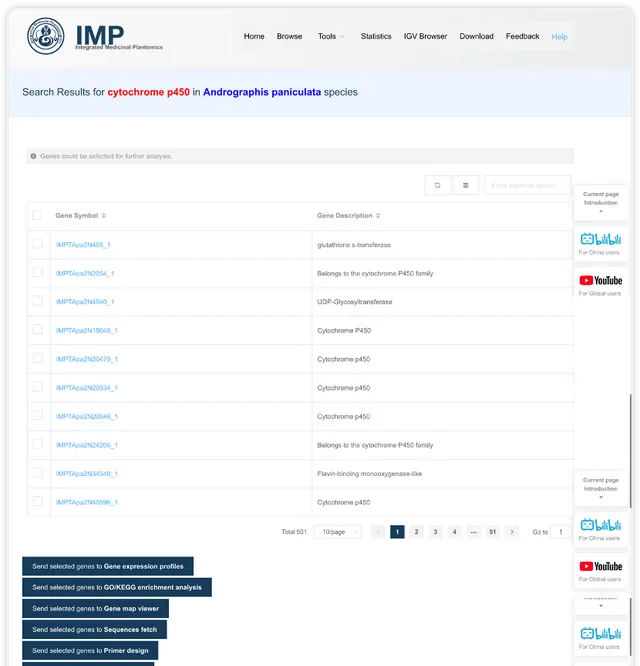
以單基因為中心的詳情頁面展示
頁面分為 3 個部份:
- 第一部份展示基因的基本資訊,包括名字資訊、功能描述資訊和序列資訊。
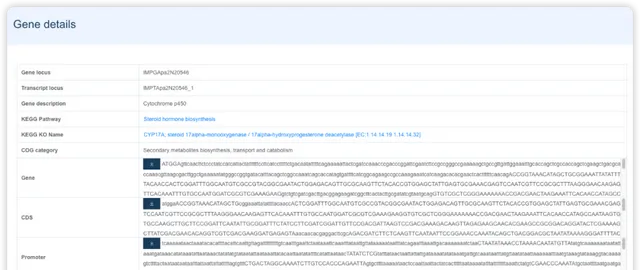
- 第二部份展示基因在不同數據集的表達圖譜資訊。
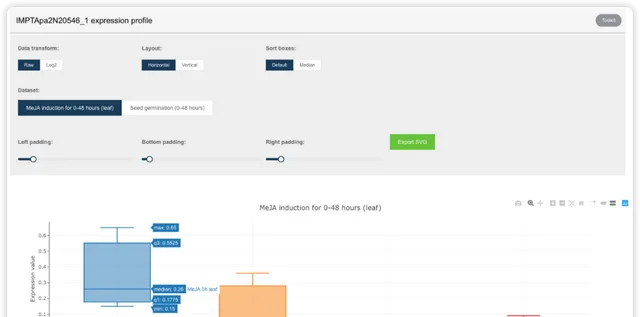
- 第三部份展示基因的結構(內含子、外顯子、UTR 等資訊)和蛋白功能域資訊。
多基因表現圖譜
可以自己按頁面選擇物種、數據集、樣品(非必選的選項如果不選,預設是全選)、輸入基因,也可以從搜尋結果中直接帶過來基因列表。
模糊搜尋:支持根據基因的功能描述關鍵詞進行模糊搜尋,獲取基因名,用於研究一類基因的表達圖譜。
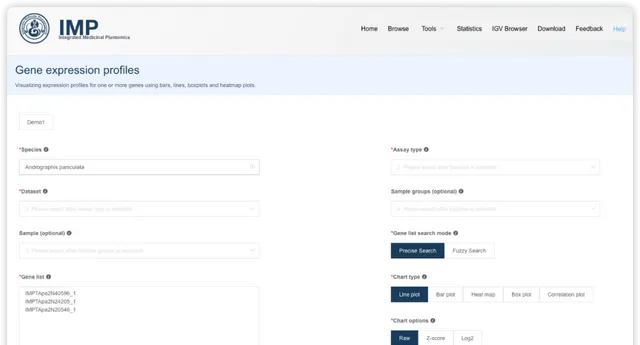
送出後獲得基因表現圖譜展示。
- 使用者可以跳轉圖形的 padding 資訊和高度資訊
- 視覺化結果可以匯出 SVG 格式
- 作圖數據可以下載,匯入 ImageGP/BIC平台進行再次分析
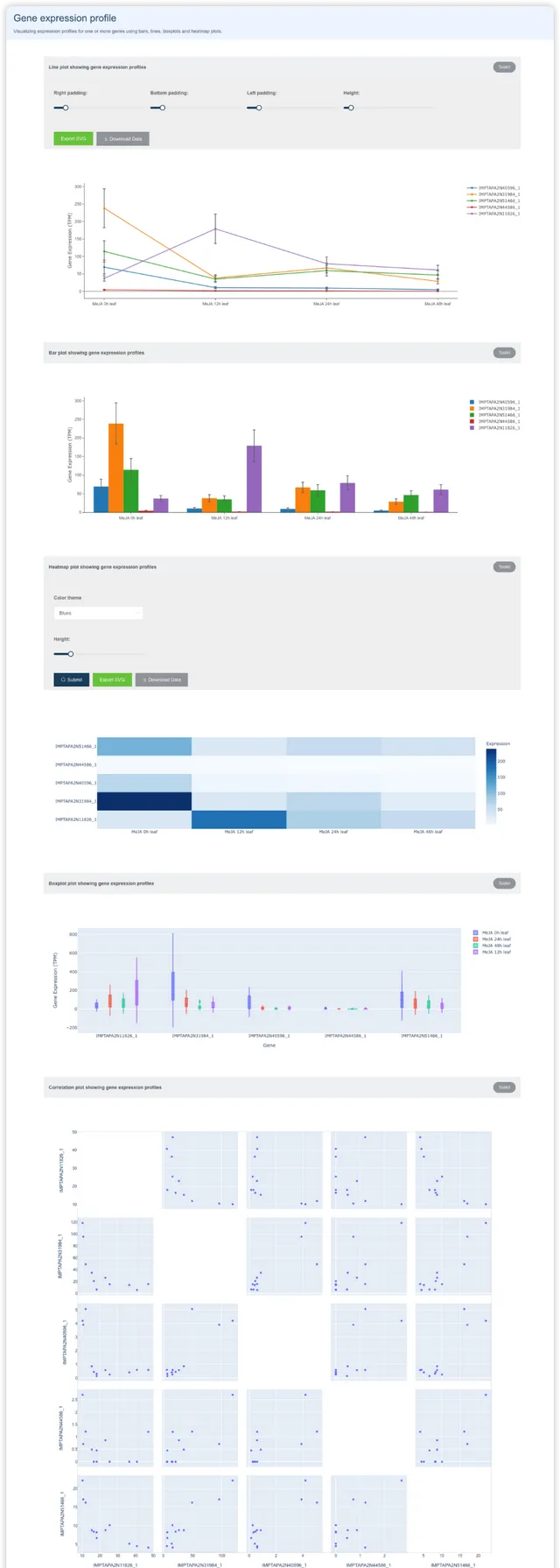
Gene fishing 調取表達模式相近的基因
選擇物種、Assay type、匹配模式,輸入基因名(可以透過Send to 功能從其它頁面發送過來),送出後獲得一個相關性網路圖和對應的結果數據。
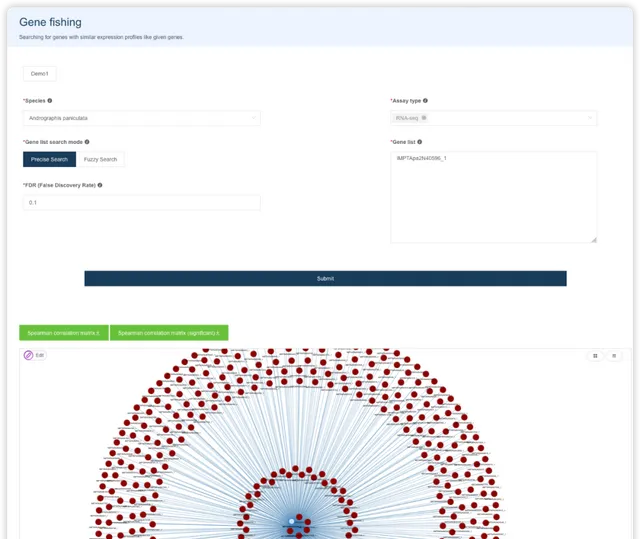
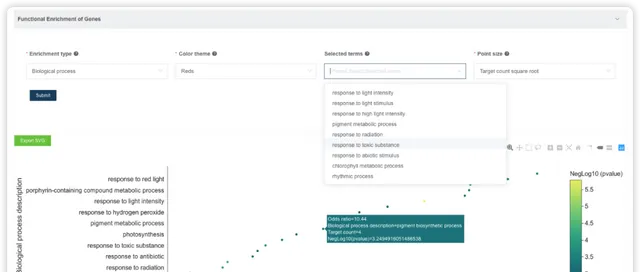
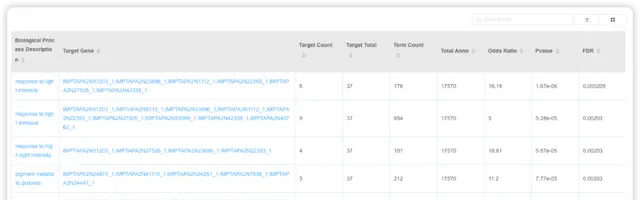
GO/KEGG 富集分析 {#gokegg}
使用者選擇物種,輸入基因名字,即可進行GO/KEGG富集分析。閱讀推文https://mp.weixin.qq.com/s/BCB16M4yI5Qa1tKyZy7WMg或檢視視訊https://www.bilibili.com/video/BV1rD4y1272a?p=4了解 GO/KEGG 富集分析的基本原理。
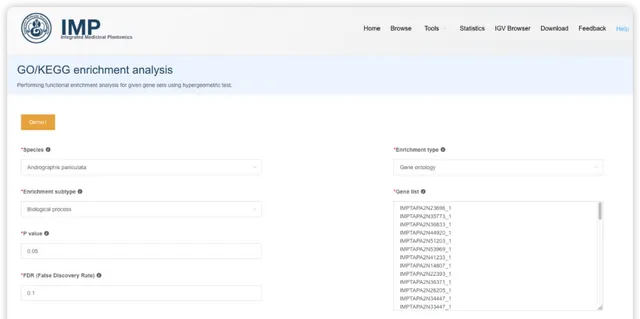
點選後,可調整富集分析結果的配色方案、選擇富集的條目進行展示。也可以下載表格檔,到高顏值免費線上繪圖平台 ImageGP/BIC https://www.bic.ac.cn/BIC進行自由繪制。
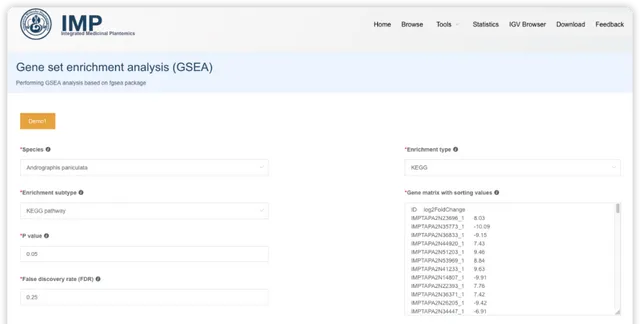
GSEA 富集分析
GSEA 富集分析的輸入會麻煩一些,目前只支持包含一列基因和一列排序值的 2 列矩陣格式;排序值可以是常見的log2(fold change) ,p-value 或也可以是其他定量值。
閱讀推文https://mp.weixin.qq.com/s/WiYUUALSmb9v5gYVxmjwjA或檢視視訊https://www.bilibili.com/video/BV1rD4y1272a?p=5了解 GSEA富集分析的輸入數據、原理和結果解讀。
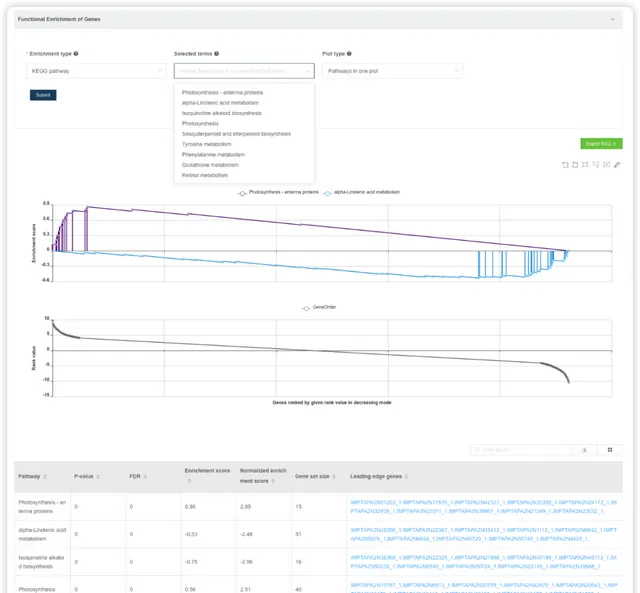
預設繪制最富集的 2 條通路在一張圖上,可以自己選擇繪制哪些通路,也可以將通路繪制在多張圖上。
BLAST序列比對和搜尋
BLAST 是鼎鼎有名的序列搜尋工具,這裏支持
非模式物種常常沒有統一的Gene Symbol ,使用的是各種意義不明的 ID,序列搜尋是把文獻或私藏的序列對映到 IMP 或在 IMP 中搜尋序列相似基因的好方法。這就是 BLAST 功能所做的。

IMP 的 Blast 功能支持使用者輸入單條或多條 FASTA 序列進行搜尋,使用者也可以選擇一個或多個或全部數據集。Advanced parameter 處可以設定更多匹配控制參數。
HTML 格式的輸出會包含匹配區域的序列比對資訊。如果使用者輸入了多條查詢序列,可在Results for 後面的下拉框中進行選擇切換。
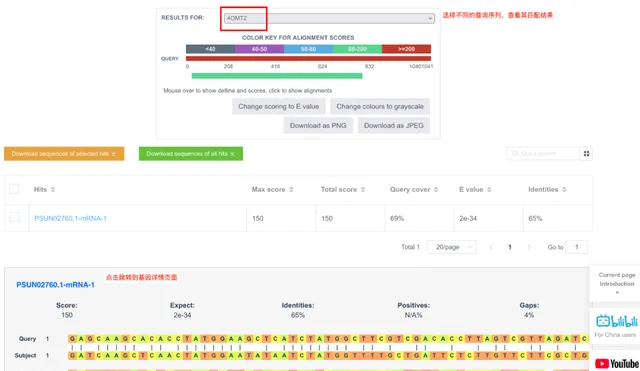
Table 格式簡潔明確地列出每條查詢序列在資料庫中的匹配序列,可以把匹配出的序列透過Send to 功能發送到更多工具頁面,快捷使用。
因為 BLAST自身的問題,如果使用者選了多個資料庫檔,當前會強制輸出 Table 格式。正在根 BLAST 溝通中,還未解決。
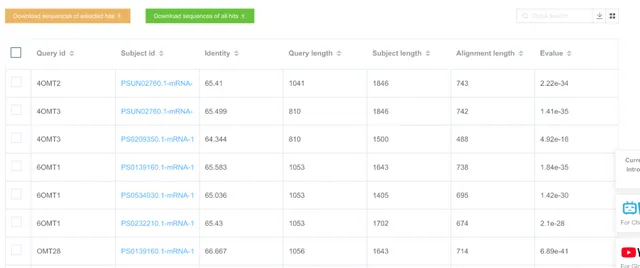
BLAST 參數參考
BLASTN 的匹配得分除以錯配罰分 (abs(reward/penalty) )的商(比值)越大表示允許的序列直接的匹配度越小。比值為 0.33 等同於序列相似度大於 99%;比值為 0.5 等同於序列相似度大於 95%;比值為 1 等同於序列相似度大於 75%。
It is important to choose reward/penalty values appropriate to the sequences being aligned with the (absolute) reward/penalty ratio increasing for more divergent sequences. A ratio of 0.33 (1/-3) is appropriate for sequences that are about 99% conserved; a ratio of 0.5 (1/-2) is best for sequences that are 95% conserved; a ratio of about one (1/-1) is best for sequences that are 75% conserved
REF: https://www.ncbi.nlm.nih.gov/books/NBK279684/
The reward/penalty values are ordered from most to least stringent, with the more stringent values better suited for alignments with high sequence identity.
|
reward/penalty |
gap costs (open/extend) |
default MegaBLAST gap costs (open/extend) |
|
1/-5 |
3/3 |
0/5.5 |
|
1/-4 |
1/2, 0/2, 2/1, 1/1 |
0/4.5 |
|
2/-7 |
2/4, 0/4, 4/2, 2/2 |
0/8 |
|
1/-3 |
2/2, 1/2, 0/2, 2/1, 1/1 |
0/3.5 |
|
2/-5 |
2/4, 0/4, 4/2, 2/2 |
0/6 |
|
1/-2 |
2/2, 1/2, 0/2, 3/1, 2/1, 1/1 |
0/2.5 |
|
2/-3 |
4/4, 2/4, 0/4, 3/3, 6/2, 5/2, 4/2, 2/2 |
0/4 |
|
3/-4 |
6/3, 5/3, 4/3, 6/2, 5/2, 4/2 |
N/A |
|
4/-5 |
6/5, 5/5, 4/5, 3/5 |
N/A |
|
1/-1 |
3/2, 2/2, 1/2, 0/2, 4/1, 3/1, 2/1 |
N/A |
|
3/-2 |
5/5 |
N/A |
|
5/-4 |
10/6, 8/6 |
N/A |
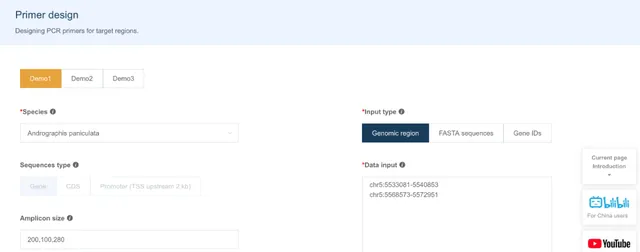
引物設計
使用者可以透過 3 種方式釘選自己的目標序列:基因組位置、序列、基因 ID,IMP 會提取對應的序列並采用 Primer3 根據設定的參數設計引物,輸出引物表格。
多序列比對展示
多序列比對是系統前進演化樹構建的字首,IMP 支持使用者直接輸入序列或提供基因名字自動提取序列進行多序列比對。
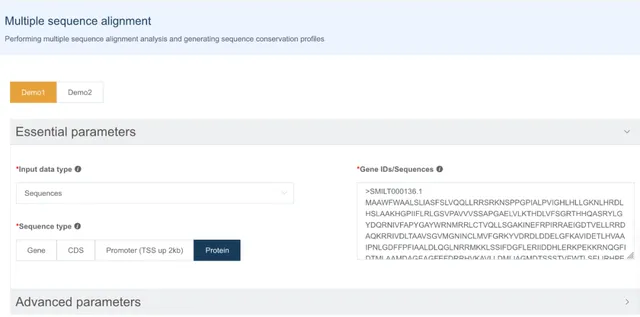
多序列比對展示處,使用者可以調整胺基酸或堿基的上色模式、一行展示的序列長度以便獲得合適長寬比的視覺化圖。

序列提取
透過功能搜尋或序列搜尋或差異基因分析完後獲得的差異基因,可貼上到這裏的Gene list 處,提取其Gene , CDS , Protein 和Promoter 序列。
基因簇視覺化
Gene map viewer 用於視覺化基因組範圍的基因分布,檢視使用者輸入的基因是否在染色體區域成簇存在。設計有 2 種展示模式:
Overlay 視覺化結果如下, 可以捲動滑鼠縮放視覺化區域,點選 Gene block 會跳轉到 IGV 頁面或基因詳情頁面:
Annotation 模式下視覺化結果如下, 如果多個基因位置在 0.2M bp內,則合並在一個三角形中展示。
更多基因共線性分析見 https://www.bic.ac.cn/SynColV 。
線上實驗設計和差異基因分析
本部份基於Reads-count 矩陣采用limma-voom 進行差異基因的鑒定,然後對篩選出的差異基因進行GO/KEGG富集分析.
該功能涉及多個分析步驟,每個步驟頁面結構差不多,下圖是對於表單部份的解釋。
第一步:實驗設計確定要比較的物種和分組資訊(不同組織部位差異或不同處理的差異)
按圖所示,順次選擇每個參數即可 (可選參數可略過)。
第二步:樣品相關性評估和過濾異常樣品
針對選中的樣品,提取其表達矩陣,並采用DESeq2 類似的方式計算量化因子獲得標準化後的數據矩陣,然後繪制樣品相關性熱圖和 PCA 分析。
下圖中的左右穿梭框顯示了系統自動鑒定出的異常樣品和透過檢測的樣品,使用者也可以根據下面的視覺化結果自行調整或篩選樣品。
下面展示的是樣品聚類熱圖和 PCA 分析的結果圖,二者都是互動式圖譜。
12個樣品的表達相關性熱圖展示。行列註釋中的DE_Group : 使用者選擇數據的生物分組資訊。Single_group : 檢查是不是有某個組只有 1 個生物學重。Single_batch : 檢查某個批次的數據是不是只有 1 個樣本。Outlier : 標識系統鑒定出的異常樣品。Suggest_remove : 建議移除的樣品。
視覺化樣品在主成分分析獲得的第 1 和 2 組成分構成的空間中的分布. Toolkit 部份使用者可以選擇其它主成分進行展示,也可以調整點的顏色、大小、形狀和繪制數據的分布模式。
第三部,設定比較組
拖動要比較的組到對應的框裏面去從而進行兩兩比較。
拖動設定比較組.
第四步:設定差異基因過濾閾值
計算出的 FDR 值低於使用者指定的值且表達變化倍數高於使用者指定的值得基因定義為差異基因。
第五步:概覽樣品資訊和設定的參數,這一步是送出前的資訊確認
送出前確認樣本資訊和參數資訊。
第六步:差異基因分析結果報告
差異基因分析結果報告包含樣品資訊、樣品相關性熱圖、PCA 分析、差異基因熱圖、差異基因火山圖、功能富集分析結果等。每一部份結果圖都可以做進一步客製,也可以匯出數據,放到一款高顏值免費線上SCI繪圖工具ImageGP做更多視覺化分析。
目錄展示結果報告整體內容,各個部份可點選直接跳轉。
第一部份是樣品整體相關性資訊展示。
第二部份是差異基因和富集分析結果展示。
整個結果也可以匯出為 PDF 格式:當所有結果完成載入後,按Ctrl + p 會啟動Printer to PDF 或打印到 PDF 功能,點選確認後即可輸出 PDF。
IGV 基因組瀏覽器 {#igvch}
IGV 瀏覽器常用與視覺化高通量數據在全基因組範圍或局部基因區域的分布,可以用於展示基因表現豐度的高低,也可以用於發現新的可變剪接事件。
關鍵資訊
所有的 track 檔都已標準化為了RPM (reads per million).
所有的 track 縱軸最大值和最小值得已設定為同一個標度,不同 track 的峰圖的高低是可比的。
支持基因名字檢索。
文章發表
IMP 於 2023 年 10 月發表於 Nucleic Acids Research , https://doi.org/10.1093/nar/gkad898。
引文:IMP: bridging the gap for medicinal plant genomics. Nucleic Acids Research, gkad898, https://doi.org/10.1093/nar/gkad898











