一、基因编辑及其原理
基因编辑又称基因组编辑或基因组工程,是一种新兴的、比较精确的能对生物体基因组特定目标基因进行修饰的基因工程技术。它 可以精确地定位到基因组的某一位点上,在该位点上可以进行特定DNA片段的插入、缺失、修改和替换。
如果我们将基因比作一个代码文件的话,DNA代表了对这个文件的「存」,基因测序工作的完成代表了对这个文件的「读」,基因编辑则代表对这个文件的「写」。因此,基因编辑仿佛是一把有魔力的「剪刀」,既可以「剪切」基因,也可以应用于「修补」基因。通过对DNA序列的改变和修正,破坏、抑制或恢复基因的功能,来实现遗传治疗、基因研究、农作物改良等等应用。
基因编辑工具使人类拥有了定向改变DNA或RNA的能力,其核心能力是精准定位与安全修改。精准定位是实现定向编辑的第一步。靶向特定目标序列,而不是在DNA任意位置随意更改,使基因编辑成为可控的工具,并且规避不受控的基因突变带来的风险。在精确定位的基础上,基因编辑还应能够识别并定位到任意的设定序列,并进行安全的修改,这样才能够针对不同特异性基因进行编辑。
为完成这两个步骤,基因编辑的工作原理利用了DNA断裂及修复过程中的特点。我们已经知道DNA是一个细长的分子,细长就意味着容易断裂(DNA双链断裂,Double Strand Break,DSB),而DNA对生命又太重要了,因此细胞自然就有极强的纠错机制将断裂的DNA再重新接好修复。具体地,修复途径主要有两种:非同源末端连接(Non- Homologous End Joining,NHEJ)和同源定向修复(Homology Directed Repair,HDR)。
HDR是指只有当细胞内存在与损伤DNA同源的DNA片段时,HDR才有可能能发生。通俗来讲就是,DNA断裂后,细胞虽不知道DNA本来的样子,但可以通过基因的备份(姐妹染色单体或同源序列)进行修复。细胞同样不知道备份的样子、位置,因此它就拿着断裂的DNA到处比对。如果找到一段DNA序列和断DNA缺口前后的序列相似到一定程度(即上面所说的同源),那细胞就会判定这是修复的备份模板,而后比照该模板进行修复。HDR主要发生在细胞的G2期和S期。
当细胞内没有相应的同源DNA片段时,细胞将利用NHEJ修复损伤。与HDR不同,NHEJ不需要修复模板或广泛的DNA合成,而是碰到断裂就直接将其修好。因此它在整个细胞周期中都是活跃的,具有更高的修复能力。
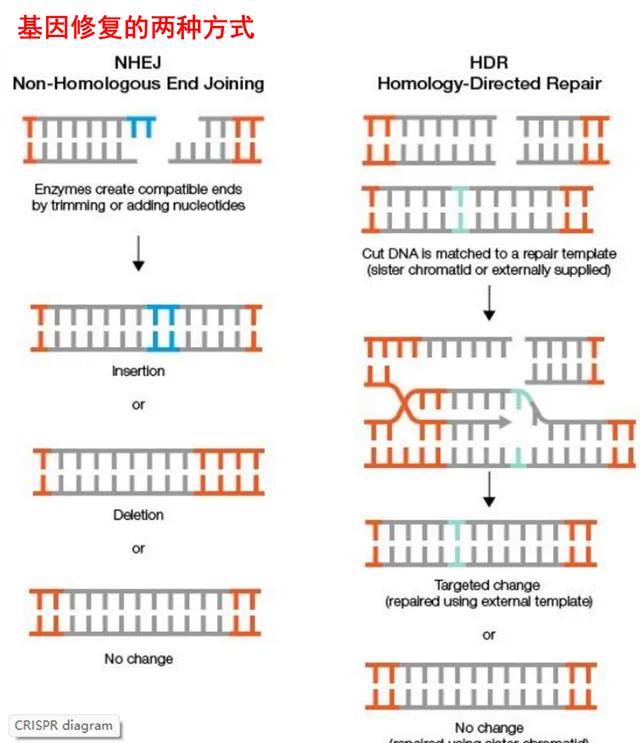
基因编辑的工作原理利用的便是这两种路径的特点。其一是诱导DNA的损伤修复并不总是能够修复的完好如初。虽然DNA纠错机制极强,然而再好的能工巧匠也不是完美的,DNA的修复也一样会出错。在无数次的修复中,有的可能修复如初,但也可能出现错误或者bug,从而导致少量遗传物质的插入或缺失,产生indel效应,进而造成移码突变。这样,该基因将产生肽链缩短且功能缺失的蛋白质,从结果上看等同于敲除了这个基因。
其二则是HDR的模板修复。由于HDR依照的模版序列可来自另一条姊妹染色体也可来自外部引入的DNA模版。因此,若以姊妹染色体作为模版序列,断裂的DNA将被恢复成原样;若以外部引入的序列作为模版,修复后的DNA将包含模版中间的序列。
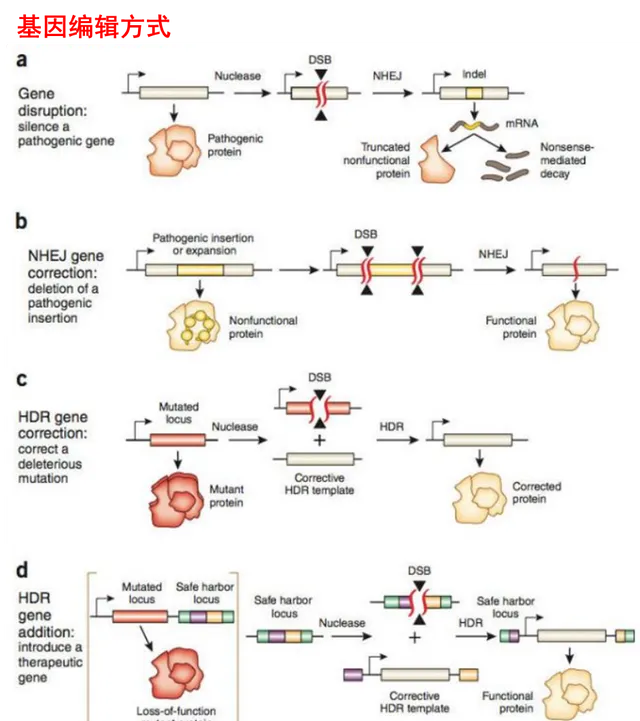
因此,如果想对目标基因实现编辑,我们就可以利用这两个特点,为目标基因设计一套系统(通俗来说就是设计、制作一把「剪刀」),完成精准定位和安全修改两个步骤就可以。通常,主要的编辑动作包括基因敲除、基因修复和基因插入。
基因敲除是指利用」剪刀「定位到目标基因点位,并在此引发DSB,而后基因会通过NHEJ进行修复,在此过程中如果被修复如初。那它就会继续被定位,就这样不断切、不断修复。直到切到DNA被修错,序列发生变化而无法被定位为止。相应地基因修复主要是针对变异的基因,使用」剪刀「精准定位变异位置,将其剪切形成DSB,而后基因通过NEHJ或HDR修复如初。基因插入则同时传入「剪刀」和插入基因模板。先通过「剪刀」引入DSB,而后基因通过HDR按照插入基因模板进行修复。
实际应用中,基因编辑的精准定位剪刀是通过序列特异性的DNA结合结构域和非特异性的DNA修饰结构域组合而成的序列特异性核酸内切酶。它可以通过特异性序列识别染色体上的DNA靶位点,进行切割并产生DSB诱导DNA的损伤修复,从而实现对指定基因组的定向编辑。
简单来说,基因编辑就是利用一个蛋白或融合蛋白作为DNA分子的剪刀对目的基因进行改造。该蛋白的一部分结构可以识别、结合要编辑DNA的特定区域(这样才能把剪刀带到需要改造的基因那里,即精准定位),一部分结构可以对DNA进行操作(这样才能发挥剪刀的功能,进行基因的定点定向改造,即安全修改)。
二、基因编辑工具
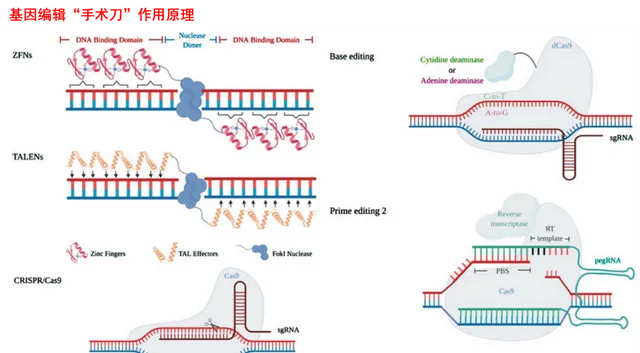
目前,基因编辑工具已从第一代的ZFNs(Zinc Figer Nucleases,锌指核酸内切酶)、第二代的TALENs(Transcription Activator-Like Effector Nucleases,类转录激活因子效应物核酸酶),发展到第三代的 CRISPR/Cas(Clustered Regularly-Interspaced Short Palindromic Repeats/CRISPR associated proteins system,成簇规律间隔短回文重复序列/CRISPR相关蛋白系统)。此外,还有基于CRISPR/Cas开发的单碱基编辑(Base Editing,BE)和先导编辑(Prime Editing,PE)等,它们已在多种生物体系中得到广泛应用。
1. ZFNs
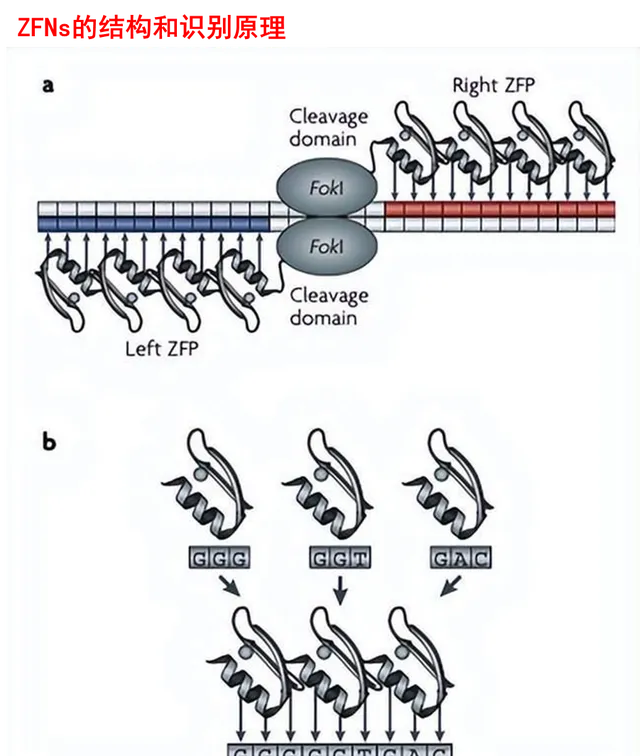
ZFNs是人工设计的含有两个功能结构域的蛋白核酸内切酶,包括决定其特异性的锌指蛋白结构域和切割DNA的Fok I核酸酶结构域,也就是说ZFNs这把「剪刀」的定位能力来自于锌指蛋白结构域,切割修改能力来自于Fok I核酸酶结构域。
锌指蛋白最早发现于非洲爪蟾的细胞,被用来定位基因组序列并启动特定基因的转录和蛋白质合成。利用其这一特性,人们可以根据想要编辑的基因人工设计锌指结构并将其与Fok I融合,由此形成第一代基因编辑工具ZFNs。
ZFNs中的锌指蛋白结构域负责DNA识别、结合,其结构决定了识别位点的特异性。锌指蛋白结构域一般包含3-6个独立的锌指重复结构,每个锌指结构能识别3个碱基,因而一个锌指结构域可以识别9-18bp长度的特异性序列。核酸内切酶Fok I则必须形成二聚体才能够将DNA双链剪开,因此需要在目标位点左右两端都设计锌指酶。
2. TALENs
TALENs也是由两部分组成:一部分是类转录激活因子样效应蛋白(Transcription Activator-Like Effector,TALE),负责对目标序列的识别与结合,提供「剪刀」的定位能力;另一部分是与ZFN相同的FokI核酸酶,提供「剪刀」的切割能力。
TALE蛋白最初是在黄单胞菌中发现的,人们发现其中心结构域的串联重复序列(Repeat domain)可以特异性识别DNA序列。利用这一特性,可以人为设计TALE蛋白作为DNA结合域,并将其与FokⅠ核酸酶偶联,即可构建第二代编辑工具TALENs。
TALENs中高度保守的33~35个氨基酸重复单元决定了TALE蛋白的识别特异性。为了切割靶DNA的两条链,必须将FokI切割结构域二聚化。因此,和锌指一样,TALENs也需要成对设计才能发挥作用。

3. CRISPR/Cas
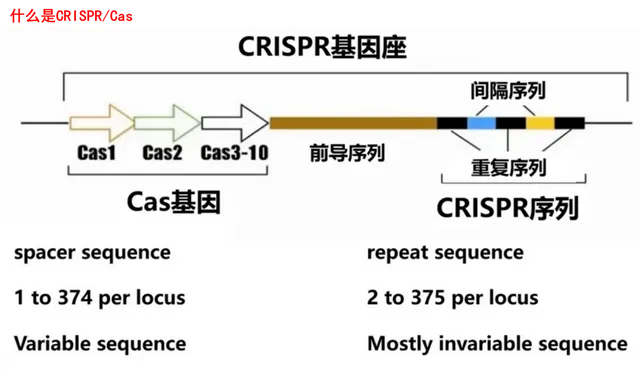
CRISPR/Cas系统,顾名思义,由CRISPR基因阵列和Cas基因(CRISPR相关蛋白基因)组成。其中CRISPR由一系列短的高度保守的正向重复序列(repeats)与长度相似的间隔序列(spacers)间隔排列组成,上游有负责转录的前导序列。CRISPR转录、切割最终形成的crRNA(CRISPR RNA),能引导对靶基因的剪切,为「剪刀」提供定位能力。
Cas基因即CRISPR相关蛋白基因,是靠近CRISPR基因座附近的一组高度保守基因群,其所编码的Cas蛋白,包含核酸内切酶、解旋酶以及与核糖核酸结合的结构域,能够切割DNA双链,为「剪刀」提供切割能力。
与前两代基因编辑工具一样,CRISPR/Cas也是先揭示了自然界中已有的机制,而后才开始人工设计的。CRISPR/Cas系统广泛存在于细菌和古细菌的基因组中,是一种细菌降解入侵的病毒DNA或其他外源DNA的免疫机制。
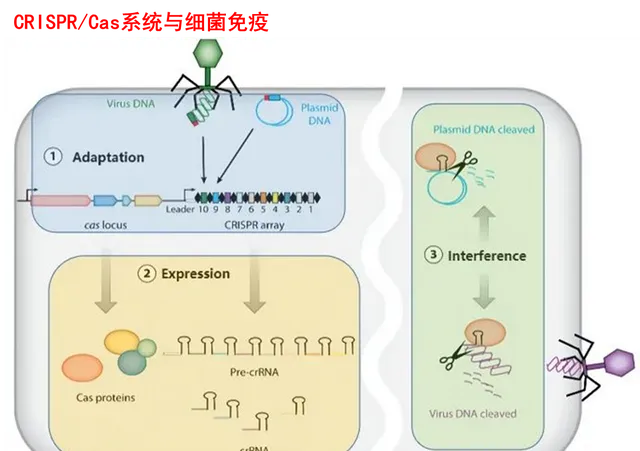
CRISPR/Cas系统进行免疫防御主要分为3个阶段:适应(Adaptation )、表达(Expression)、干扰(Interference),这与真核生物中RNA干扰(RNAi)的原理是相似的。
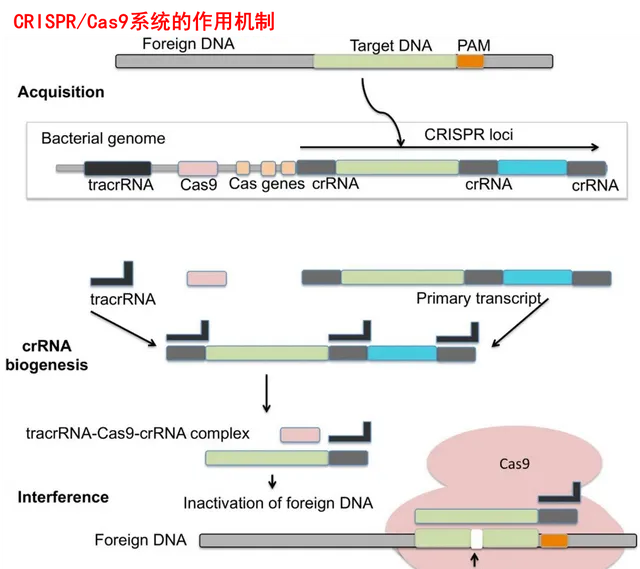
首次入侵的外源DNA,Cas编码的Cas蛋白会通过PAM(Protospacer Adjacent Motif,前间隔序列邻近基序) 识别出其临近的前间隔序列(protospacer),并将其切除整合到CRISPR序列中,成为其中的一段spacer,使系统拥有获得性免疫及遗传基因,此即为适应阶段。形象类比的话,就像CRISPR/Cas系统根据PAM这一身份证对外源DNA画像并记在自己的小本本上。
注:为何识别PAM作为身份证呢?直接识别前间隔序列它不香吗?仔细想想,实际上如果识别序列本身的话,细菌剪切插入之后的间隔序列就是细菌基因组的一部分了,那这时候Cas蛋白切不切自己呢?显然细菌不可能切自己,它需要区分自身和外来的间隔序列,这时候PAM就是最好的身份证了。
当外源DNA再次入侵时,细菌就开始拿出小本本,启动CRISPR序列的转录,形成一条包含spacers和repeats转录出的RNA,即前crRNA序列(pre-crRNA)。而后再根据入侵者的类型,选取对应的「身份证号码」,由核糖核酸酶或编码的Cas蛋白在repeats位点内剪切成能与目的基因碱基互补的成熟crRNA,此即为表达阶段。
前期所得的crRNA与Cas蛋白组成核酸酶复合物,扫描整个外源DNA序列,并识别出与crRNA互补的原间隔序列,定位到PAM/原间隔序列的区域,引导Cas蛋白去剪切目的基因中的基因,从而中断外源基因的表达,达到免疫防御的目的,此即为干扰阶段。
清楚了这套免疫机制的工作原理,人类就可以设计新的基因编辑「剪刀」CRISPR/Cas系统了。 即按照这个原理,就可以根据所要编辑的DNA靶标,人工设计段向导RNA。再把编码这段向导RNA和CAS蛋白DNA,用转基因的方法导入到细胞内。细胞就会制造出一大堆CAS蛋白,在向导RNA的引导下去精确切割想要编辑的那段DNA。
此外,Cas蛋白有多种类型,不同类型的Cas蛋白在其自身活性、识别位点、切割未端、RNA需求等方面具有不同的特性。因此人们还可以在自然界寻找、发现新的Cas蛋白,或者通过对Cas蛋白设计突变、定向进化产生新的Cas蛋白,以改进CRISPR/Cas系统。
根据Cas蛋白的组成和效应复合物的性质,CRISPR/Cas系统分为 class1和 class2两个大类。其中 class1的效应复合物由多个(一般4-7个)Cas蛋白亚基组成,大家一起上才能完成目标;而 class2的效应复合物只需一个多结构域蛋白,如Cas9等,一个Cas蛋白就能搞定。
此外,根据剪切模块的序列和功能特征,以及辅助模块的构成, class1系统又可进一步分为TypeⅠ、Ⅲ、Ⅳ, class2系统可进一步分为TypeⅡ、Ⅴ、Ⅵ。Makarova等人在2011年第一次对CRISPR/Cas系统的进化分类进行了整理。
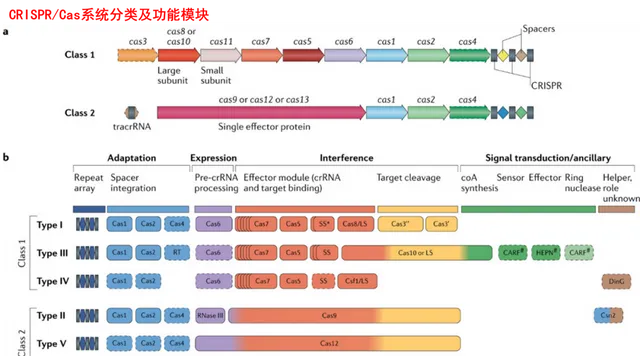
此后随着越来越多的Cas蛋白出现,CRISPR/Cas系统分类于2015年和2020年分别进行了更新。在2020年最新的分类规则中, class 1扩展到3个类型和16种亚型。 class 2扩展到3个类型和17个亚型,这些新发现包括多种V型和VI型系统。
上面已经说到两个CRISPR/Cas系统中, class 2只需要一个RNA引导的Cas核酸酶就能够完成对靶点的切割,因此使用起来更为简单、方便,有效驱动了研究者在此类中寻找潜在的新基因组编辑和诊断工具。
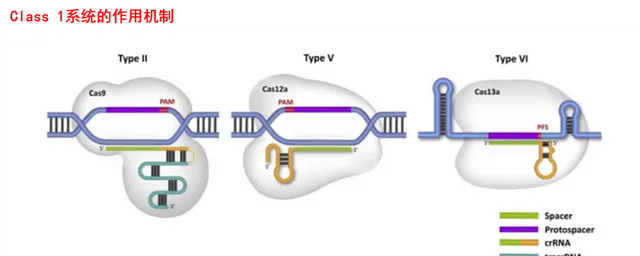
这其中CRISPR/Cas9是第一个被设计用于人类细胞基因编辑的系统,来自化脓链球菌,其系统构成简单、特异性优良、切割效率高,因此也是现在最广泛使用的CRISPR基因编辑器。
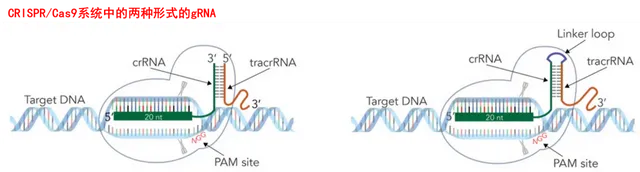
与Cas12和Cas13的工作原理不同的是,除了产生crRNA外,Cas9蛋白上游的互补链还会转录tracrRNA(transactiving CRISPR RNA,转录激活crRNA),tracrRNA具有一段能同crRNA上的重复序列互补配对的序列。两者通过碱基配对形成gRNA(guide RNA,向导RNA),指导Cas蛋白在目标DNA上引起双链断裂。
由成熟的crRNA与tracrRNA形成的gRNA是双链RNA结构。实操中为了简化操作,研究者将crRNA和tracrRNA使用linker连接,即构成了单向导RNA(single guide RNA ,sgRNA),其作为向导更简便、更经济。
4. BE
BE是基于CRISPR/Cas9技术开发的基因编辑工具,与上述基因编辑工具都是依赖于DNA双链断裂之后的HDR和NHEJ进行修复不同,BE提供了不引入DSB和外源DNA模板条件下,就可以对单个碱基进行转换的可能性。
BE目前主要有两种,一种是胞嘧啶碱基编辑器(Cytosine Base Editors,CBEs),可以将C-G变成T-A,另一种是腺嘌呤碱基编辑器(Adenine Base Editors,ABEs),可以将A-T转变成G-C。
具体来说,BE系统主要由Cas9切口酶(Cas9n)、sgDNA和脱氨酶组成。BE使用的Cas9n是失活或部分失活处理的,但其可与目标DNA结合,使得sgDNA与目标序列的互补链结合,打开DNA双螺旋,此时由脱氨酶对靶位上的单个碱基进行转变。而后,两条DNA链碱基不配对,互补链经过DNA修复之后与新碱基配对,这样就实现了单一碱基对的替换。
因此,BE系统这把「剪刀」的定位能力仍是由sgDNA提供的,修改能力则是由Cas9n和脱氨酶提供的。
注:Cas9的核酸酶剪切活性取决于两个结构域:RuvC和HNH,它们分别负责切割DNA链的两条链,且能单独被人工点突变失活。突变包括RuvC催化结构域的第10位天冬氨酸突变为丙氨酸(D10A)以及HNH催化结构域的第840位组氨酸突变为丙氨酸(H840A)。
前者发生则产生Cas9 D10A突变体,呈现RuvC失活(RuvC-)、HNH(HNH+)激活的状态;后者发生则产生Cas9 H840A突变体,呈现RuvC激活(RuvC+) 、HNH失活(HNH-)的状态。两种突变体的Cas9仍然具有核酸酶活性,可对靶向序列进行单链剪切。
两者同时发生,RuvC和HNH同时处于失活状态时(D10A&H840A; RuvC-&HNH-),Cas9将不具有核酸酶活性,成为dCas9(dead Cas9)。dCas9虽然没有剪切DNA的能力,但仍然可以在gRNA的引导下与特定的DNA序列结合。
对于CBEs系统,当Cas9n-胞嘧啶脱氨酶融合蛋白在sgRNA的引导下靶向基因组DNA时,胞嘧啶脱氨酶可结合到由Cas9n、sgRNA及基因组DNA形成的R-loop区的ssDNA(Single-Stranded DNA,单链DNA)处,将该处一定范围内的C脱氨为U,进而通过DNA修复或复制将U转变为T,最终实现C-T至G-A的转换。
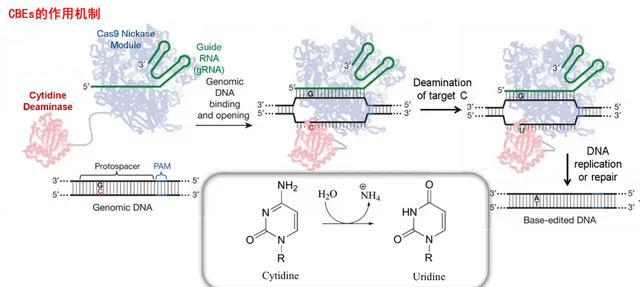
对于ABEs系统,当Cas9n-腺嘌呤脱氨酶 融合蛋白在sgRNA的引导下靶向基因组DNA时,腺嘌呤脱氨酶 可结合到由Cas9n、sgRNA及基因组DNA形成的R-loop区的ssDNA处,将该处一定范围内的A脱氨为肌苷(I),肌苷在DNA水平会被当作G进行读码与复制,最终实现A-T至G-C的转换。
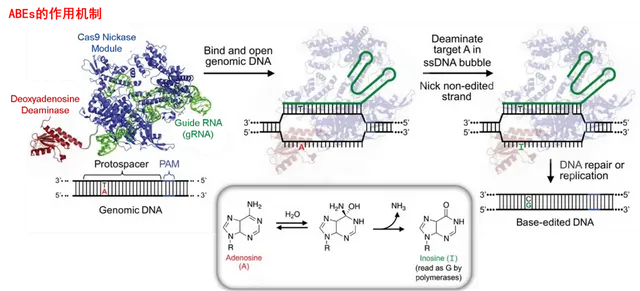
5. PE
PE是基于BE技术开发的基因编辑工具,简单来说就是在BE技术的基础上引入逆转录的方法,不仅可以实现单个碱基转换,还可以实现碱基敲除、添加等多种操作。
PE系统主要由三个部分组成:Cas9 H840A切口酶、逆转录酶(Reverse transcripatse,又称反转录酶,RT酶) ,以及起引导作用的pegRNA(prime editing guide RNA,PE向导RNA)。
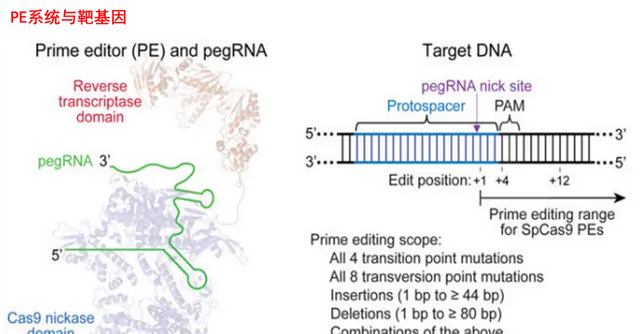
PE的作用机制是pegRNA的引导偶联的Cas9 H840A和RT酶 靶向结合并切开一条DNA链,而后逆转录生成的单链DNA会在该切口处与原始序列展开竞争,由于DNA修复的酶一般从5端结合DNA链,所以原始的链倾向于被清除掉,希望加入的片段则被优先整合到原序列中。这样即可实现碱基的替换、增减,以及DNA片段的增减。
因此,PE系统这把「剪刀」的定位能力是由pegDNA提供的,修改能力则是由偶联的Cas9 H840A和RT酶 提供的。

三、不同基因编辑工具的比较
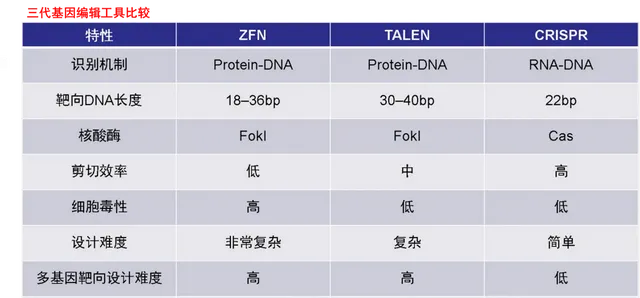
基因编辑工具的进化中,最重要的一点是为剪刀提供定位功能模块的变化。在ZFN之前,其实还有一种称为兆核酸酶(Meganuclease)的基因编辑技术。
由于对DNA序列的识别位点较大(>14 bp),兆核酸酶特异性高,且其自身编码基因较小(仅1 Kb左右)易于传递。然而,兆核酸酶的DNA结合域和切割域不易区分,设计困难;一旦更换靶基因就要重新设计,做大规模蛋白质工程,成本较高。且多数兆核酸酶很难在人基因组上找到合适的位点,使其在一定程度上应用受限。
ZFN和TALEN把核酸酶的识别域和切割域分开,因此换靶基因时只需定制识别域就行(即只需做针对识别域的蛋白质工程)。不同的是,TALEN蛋白结构具有重复氨基酸序列,相比ZFN锌指结构域的设计挑战较小,且靶向特异性更高,但TALEN在size上增大了递送难度(~6kb vs.~2kb)。
到了CRIPSR/Cas系统,终于不需要折腾蛋白才能更改靶基因了,只需定制一个sgRNA就能靶向目标,设计难度、构建难度、综合成本大大降低。此外,CRISPR/Cas的识别精度、剪切效率也更高。
脱靶效应和细胞毒性方面,由于ZFN通过二聚体产生作用,一旦形成异源二聚体,就很可能造成脱靶效应,TALEN也一样容易产生脱靶效应,只不过细胞毒性比ZFN要小。CRISPR/Cas的脱靶效应和细胞毒性都较低。
上述三代基因编辑工具的底层原理都是先引发DNA的DSB,再利用NHEJ或HDR对DSB进行修复。这会产生两个问题。
一是两种修复途径中NHEJ总是占优势的,很容易在基因敲入中引发基因敲除。因此在基因敲入中需要考虑如何增加HDR的概率(如CRISPR/Cas9各改进款式中,或者加NHEJ抑制剂从而减少NHEJ的占比,或者加HDR促进剂从而拉高HDR的占比)。此外,HDR发生在细胞分裂期,对于不分裂的细胞(如神经系统)很难实现基因敲入。
二是DSB可能是基因组中最严重的事故之一,这也是为什么细胞会不惜错误也要用NHEJ紧急修复它。DSB带来的风险包括基因组不稳定、片段改变、染色体易位以及致癌风险等。引发DSB意味着引入风险。
在这种情况下,不依赖DSB修复机制的基因编辑技术BE和PE应运而生。BE技术具有编辑效率高、编辑损伤少等特点,但是使用脱氨酶可能增加癌变的风险,而且可能出现的脱靶效应会严重影响编辑效率。
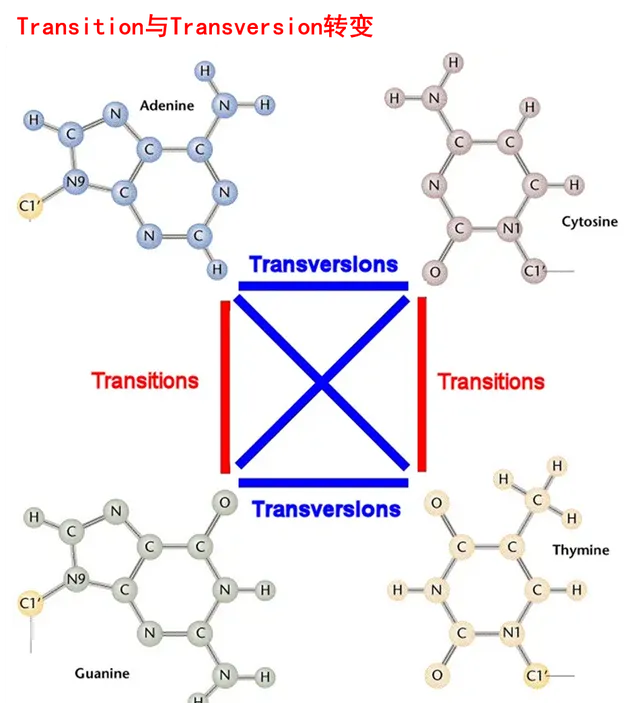
此外,BE只能做到嘌呤-嘌呤或者嘧啶-嘧啶的互相转变(即transition),对嘌呤-嘧啶的互相转变(即transversion)则无能为力。
PE则针对BE的脱靶现象进行了改进,不仅较好解决了脱靶的问题,而且可以同时实现transition和transversion,也就是说PE可以实现全部12中碱基的转换。只不过PE技术还不成熟,其可靠性有待进一步研究,而且与BE技术相似,使用逆转录酶和脱氨酶仍然存在安全隐患。
至此,我们已经对基因编辑的原理、基因编辑工具及其对比有了清晰的了解。新技术的诞生与发展,不仅使其作为更精准、更高效的基因研究工具被开发,也因其在基因筛选、模型构建、机制研究中发挥了不可替代的独特作用,为疾病治疗提供了新思路、新范式,当然也为我们的侦察兵章鱼托波的诞生提供巨大助力。
温馨提示:本文仅是笔者思考内容的记录,仅供读者参考,不作为任何投资建议;投资有风险,入市需谨慎。











