一、基因編輯及其原理
基因編輯又稱基因組編輯或基因組工程,是一種新興的、比較精確的能對生物體基因組特定目標基因進行修飾的基因工程技術。它 可以精確地定位到基因組的某一位點上,在該位點上可以進行特定DNA片段的插入、缺失、修改和替換。
如果我們將基因比作一個程式碼檔的話,DNA代表了對這個檔的「存」,基因測序工作的完成代表了對這個檔的「讀」,基因編輯則代表對這個檔的「寫」。因此,基因編輯仿佛是一把有魔力的「剪刀」,既可以「剪下」基因,也可以套用於「修補」基因。透過對DNA序列的改變和修正,破壞、抑制或恢復基因的功能,來實作遺傳治療、基因研究、農作物改良等等套用。
基因編輯工具使人類擁有了定向改變DNA或RNA的能力,其核心能力是精準定位與安全修改。精準定位是實作定向編輯的第一步。靶向特定目標序列,而不是在DNA任意位置隨意更改,使基因編輯成為可控的工具,並且規避不受控的基因突變帶來的風險。在精確定位的基礎上,基因編輯還應能夠辨識並定位到任意的設定序列,並進行安全的修改,這樣才能夠針對不同特異性基因進行編輯。
為完成這兩個步驟,基因編輯的工作原理利用了DNA斷裂及修復過程中的特點。我們已經知道DNA是一個細長的分子,細長就意味著容易斷裂(DNA雙鏈斷裂,Double Strand Break,DSB),而DNA對生命又太重要了,因此細胞自然就有極強的糾錯機制將斷裂的DNA再重新接好修復。具體地,修復途徑主要有兩種:非同源末端連線(Non- Homologous End Joining,NHEJ)和同源定向修復(Homology Directed Repair,HDR)。
HDR是指只有當細胞記憶體在與損傷DNA同源的DNA片段時,HDR才有可能能發生。通俗來講就是,DNA斷裂後,細胞雖不知道DNA本來的樣子,但可以透過基因的備份(姐妹染色單體或同源序列)進行修復。細胞同樣不知道備份的樣子、位置,因此它就拿著斷裂的DNA到處比對。如果找到一段DNA序列和斷DNA缺口前後的序列相似到一定程度(即上面所說的同源),那細胞就會判定這是修復的備份樣版,而後比照該樣版進行修復。HDR主要發生在細胞的G2期和S期。
當細胞內沒有相應的同源DNA片段時,細胞將利用NHEJ修復損傷。與HDR不同,NHEJ不需要修復樣版或廣泛的DNA合成,而是碰到斷裂就直接將其修好。因此它在整個細胞周期中都是活躍的,具有更高的修復能力。
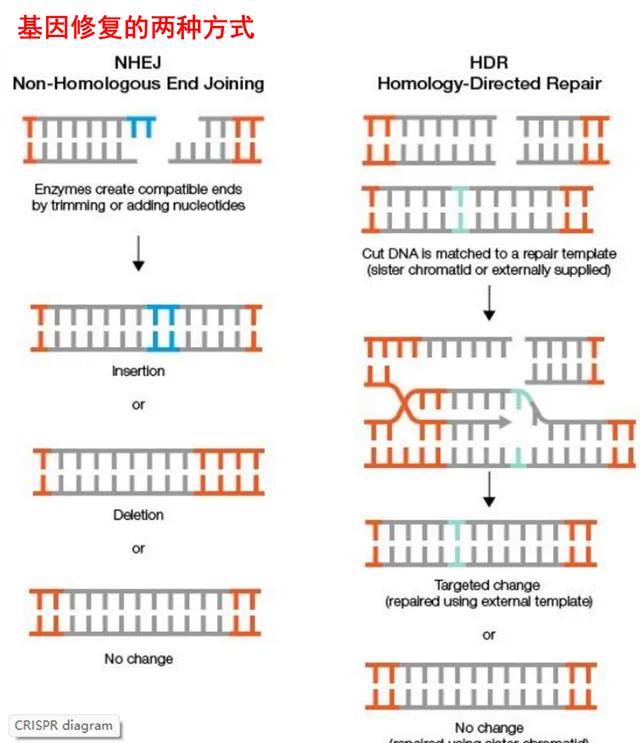
基因編輯的工作原理利用的便是這兩種路徑的特點。其一是誘導DNA的損傷修復並不總是能夠修復的完好如初。雖然DNA糾錯機制極強,然而再好的能工巧匠也不是完美的,DNA的修復也一樣會出錯。在無數次的修復中,有的可能修復如初,但也可能出現錯誤或者bug,從而導致少量遺傳物質的插入或缺失,產生indel效應,進而造成移碼突變。這樣,該基因將產生肽鏈縮短且功能缺失的蛋白質,從結果上看等同於敲除了這個基因。
其二則是HDR的樣版修復。由於HDR依照的模版序列可來自另一條姊妹染色體也可來自外部引入的DNA模版。因此,若以姊妹染色體作為模版序列,斷裂的DNA將被恢復成原樣;若以外部引入的序列作為模版,修復後的DNA將包含模版中間的序列。
因此,如果想對目標基因實作編輯,我們就可以利用這兩個特點,為目標基因設計一套系統(通俗來說就是設計、制作一把「剪刀」),完成精準定位和安全修改兩個步驟就可以。通常,主要的編輯動作包括基因剔除、基因修復和基因插入。
基因剔除是指利用」剪刀「定位到目標基因點位,並在此引發DSB,而後基因會透過NHEJ進行修復,在此過程中如果被修復如初。那它就會繼續被定位,就這樣不斷切、不斷修復。直到切到DNA被修錯,序列發生變化而無法被定位為止。相應地基因修復主要是針對變異的基因,使用」剪刀「精準定位變異位置,將其剪下形成DSB,而後基因透過NEHJ或HDR修復如初。基因插入則同時傳入「剪刀」和插入基因樣版。先透過「剪刀」引入DSB,而後基因透過HDR按照插入基因樣版進行修復。
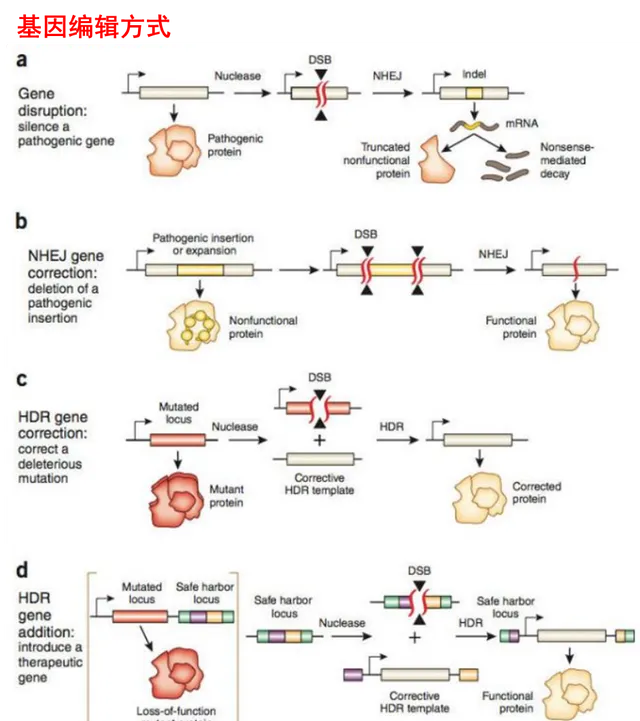
實際套用中,基因編輯的精準定位剪刀是透過序列特異性的DNA結合結構域和非特異性的DNA修飾結構域組合而成的序列特異性核酸內切酶。它可以透過特異性序列辨識染色體上的DNA靶位點,進行切割並產生DSB誘導DNA的損傷修復,從而實作對指定基因組的定向編輯。
簡單來說,基因編輯就是利用一個蛋白或融合蛋白作為DNA分子的剪刀對目的基因進行改造。該蛋白的一部份結構可以辨識、結合要編輯DNA的特定區域(這樣才能把剪刀帶到需要改造的基因那裏,即精準定位),一部份結構可以對DNA進行操作(這樣才能發揮剪刀的功能,進行基因的定點定向改造,即安全修改)。
二、基因編輯工具
目前,基因編輯工具已從第一代的ZFNs(Zinc Figer Nucleases,鋅指核酸內切酶)、第二代的TALENs(Transcription Activator-Like Effector Nucleases,類轉錄啟用因子效應物核酸酶),發展到第三代的 CRISPR/Cas(Clustered Regularly-Interspaced Short Palindromic Repeats/CRISPR associated proteins system,成簇規律間隔短回文重復序列/CRISPR相關蛋白系統)。此外,還有基於CRISPR/Cas開發的單堿基編輯(Base Editing,BE)和先導編輯(Prime Editing,PE)等,它們已在多種生物體系中得到廣泛套用。
1. ZFNs
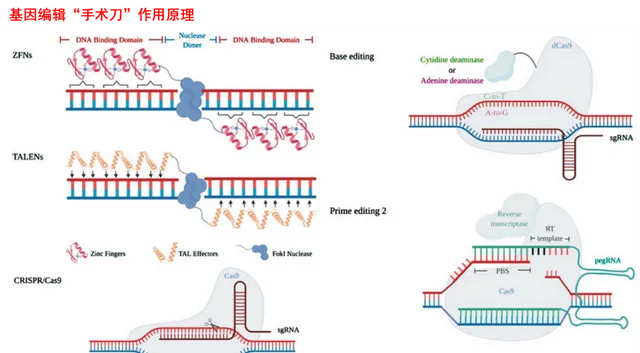
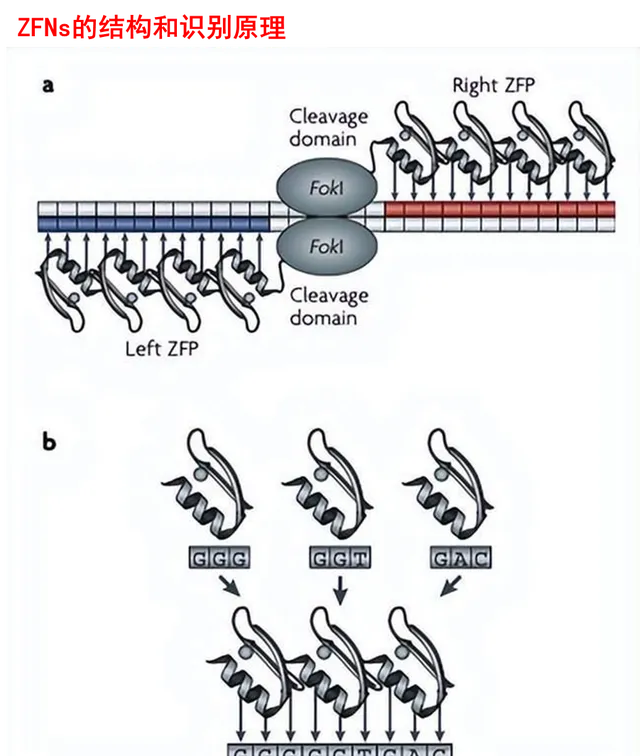
ZFNs是人工設計的含有兩個功能結構域的蛋白核酸內切酶,包括決定其特異性的鋅指蛋白結構域和切割DNA的Fok I核酸酶結構域,也就是說ZFNs這把「剪刀」的定位能力來自於鋅指蛋白結構域,切割修改能力來自於Fok I核酸酶結構域。
鋅指蛋白最早發現於非洲爪蟾的細胞,被用來定位基因組序列並啟動特定基因的轉錄和蛋白質合成。利用其這一特性,人們可以根據想要編輯的基因人工設計鋅指結構並將其與Fok I融合,由此形成第一代基因編輯工具ZFNs。
ZFNs中的鋅指蛋白結構域負責DNA辨識、結合,其結構決定了辨識位點的特異性。鋅指蛋白結構域一般包含3-6個獨立的鋅指重複結構,每個鋅指結構能辨識3個堿基,因而一個鋅指結構域可以辨識9-18bp長度的特異性序列。核酸內切酶Fok I則必須形成二聚體才能夠將DNA雙鏈剪開,因此需要在目標位點左右兩端都設計鋅指酶。
2. TALENs
TALENs也是由兩部份組成:一部份是類轉錄啟用因子樣效應蛋白(Transcription Activator-Like Effector,TALE),負責對目標序列的辨識與結合,提供「剪刀」的定位能力;另一部份是與ZFN相同的FokI核酸酶,提供「剪刀」的切割能力。
TALE蛋白最初是在黃單胞菌中發現的,人們發現其中心結構域的串聯重復序列(Repeat domain)可以特異性辨識DNA序列。利用這一特性,可以人為設計TALE蛋白作為DNA結合域,並將其與FokⅠ核酸酶偶聯,即可構建第二代編輯工具TALENs。
TALENs中高度保守的33~35個胺基酸重復單元決定了TALE蛋白的辨識特異性。為了切割靶DNA的兩條鏈,必須將FokI切割結構域二聚化。因此,和鋅指一樣,TALENs也需要成對設計才能發揮作用。

3. CRISPR/Cas
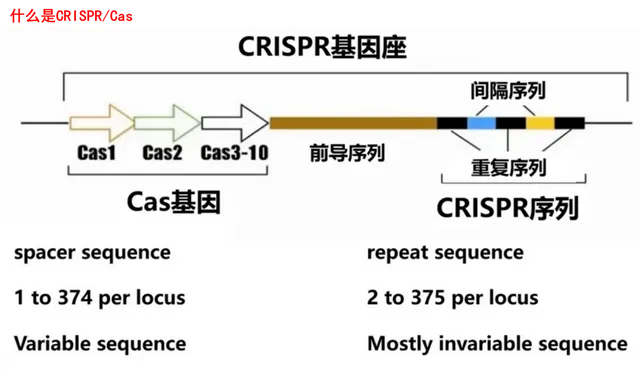
CRISPR/Cas系統,顧名思義,由CRISPR基因陣列和Cas基因(CRISPR相關蛋白基因)組成。其中CRISPR由一系列短的高度保守的正向重復序列(repeats)與長度相似的間隔序列(spacers)間隔排列組成,上遊有負責轉錄的前導序列。CRISPR轉錄、切割最終形成的crRNA(CRISPR RNA),能引導對靶基因的剪下,為「剪刀」提供定位能力。
Cas基因即CRISPR相關蛋白基因,是靠近CRISPR基因座附近的一組高度保守基因群,其所編碼的Cas蛋白,包含核酸內切酶、解旋酶以及與核糖核酸結合的結構域,能夠切割DNA雙鏈,為「剪刀」提供切割能力。
與前兩代基因編輯工具一樣,CRISPR/Cas也是先揭示了自然界中已有的機制,而後才開始人工設計的。CRISPR/Cas系統廣泛存在於細菌和古細菌的基因組中,是一種細菌降解入侵的病毒DNA或其他外源DNA的免疫機制。
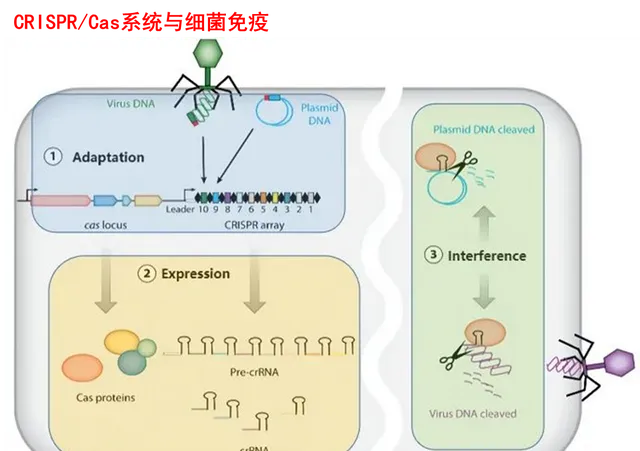
CRISPR/Cas系統進行免疫防禦主要分為3個階段:適應(Adaptation )、表達(Expression)、幹擾(Interference),這與真核生物中RNA幹擾(RNAi)的原理是相似的。
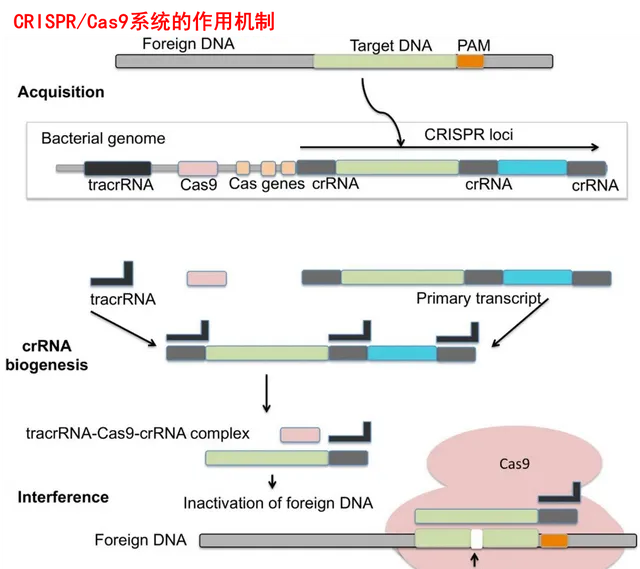
首次入侵的外源DNA,Cas編碼的Cas蛋白會透過PAM(Protospacer Adjacent Motif,前間隔序列鄰近基序) 辨識出其臨近的前間隔序列(protospacer),並將其切除整合到CRISPR序列中,成為其中的一段spacer,使系統擁有後天性免疫及遺傳基因,此即為適應階段。形象類比的話,就像CRISPR/Cas系統根據PAM這一身份證對外源DNA畫像並記在自己的小本本上。
註:為何辨識PAM作為身份證呢?直接辨識前間隔序列它不香嗎?仔細想想,實際上如果辨識序列本身的話,細菌剪下插入之後的間隔序列就是細菌基因組的一部份了,那這時候Cas蛋白切不切自己呢?顯然細菌不可能切自己,它需要區分自身和外來的間隔序列,這時候PAM就是最好的身份證了。
當外源DNA再次入侵時,細菌就開始拿出小本本,啟動CRISPR序列的轉錄,形成一條包含spacers和repeats轉錄出的RNA,即前crRNA序列(pre-crRNA)。而後再根據入侵者的類別,選取對應的「身份證號碼」,由核糖核酸酶或編碼的Cas蛋白在repeats位點內剪下成能與目的基因堿基互補的成熟crRNA,此即為表達階段。
前期所得的crRNA與Cas蛋白組成核酸酶復合物,掃描整個外源DNA序列,並辨識出與crRNA互補的原間隔序列,定位到PAM/原間隔序列的區域,引導Cas蛋白去剪下目的基因中的基因,從而中斷外源基因的表達,達到免疫防禦的目的,此即為幹擾階段。
清楚了這套免疫機制的工作原理,人類就可以設計新的基因編輯「剪刀」CRISPR/Cas系統了。 即按照這個原理,就可以根據所要編輯的DNA靶標,人工設計段精靈RNA。再把編碼這段精靈RNA和CAS蛋白DNA,用轉基因的方法匯入到細胞內。細胞就會制造出一大堆CAS蛋白,在精靈RNA的引導下去精確切割想要編輯的那段DNA。
此外,Cas蛋白有多種類別,不同類別的Cas蛋白在其自身活性、辨識位點、切割未端、RNA需求等方面具有不同的特性。因此人們還可以在自然界尋找、發現新的Cas蛋白,或者透過對Cas蛋白設計突變、定向前進演化產生新的Cas蛋白,以改進CRISPR/Cas系統。
根據Cas蛋白的組成和效應復合物的性質,CRISPR/Cas系統分為 class1和 class2兩個大類。其中 class1的效應復合物由多個(一般4-7個)Cas蛋白亞基組成,大家一起上才能完成目標;而 class2的效應復合物只需一個多結構域蛋白,如Cas9等,一個Cas蛋白就能搞定。
此外,根據剪下模組的序列和功能特征,以及輔助模組的構成, class1系統又可進一步分為TypeⅠ、Ⅲ、Ⅳ, class2系統可進一步分為TypeⅡ、Ⅴ、Ⅵ。Makarova等人在2011年第一次對CRISPR/Cas系統的前進演化分類進行了整理。
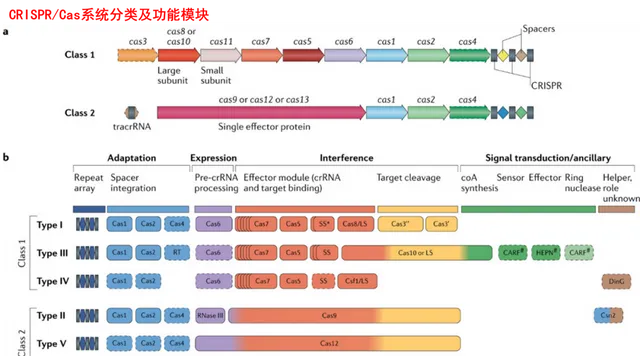
此後隨著越來越多的Cas蛋白出現,CRISPR/Cas系統分類於2015年和2020年分別進行了更新。在2020年最新的分類規則中, class 1擴充套件到3個類別和16種亞型。 class 2擴充套件到3個類別和17個亞型,這些新發現包括多種V型和VI型系統。
上面已經說到兩個CRISPR/Cas系統中, class 2只需要一個RNA引導的Cas核酸酶就能夠完成對靶點的切割,因此使用起來更為簡單、方便,有效驅動了研究者在此類中尋找潛在的新基因組編輯和診斷工具。
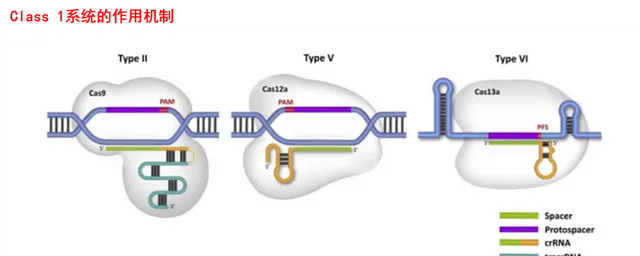
這其中CRISPR/Cas9是第一個被設計用於人類細胞基因編輯的系統,來自化膿鏈球菌,其系統構成簡單、特異性優良、切割效率高,因此也是現在最廣泛使用的CRISPR基因編輯器。
與Cas12和Cas13的工作原理不同的是,除了產生crRNA外,Cas9蛋白上遊的互補鏈還會轉錄tracrRNA(transactiving CRISPR RNA,轉錄啟用crRNA),tracrRNA具有一段能同crRNA上的重復序列互補配對的序列。兩者透過堿基配對形成gRNA(guide RNA,精靈RNA),指導Cas蛋白在目標DNA上引起雙鏈斷裂。
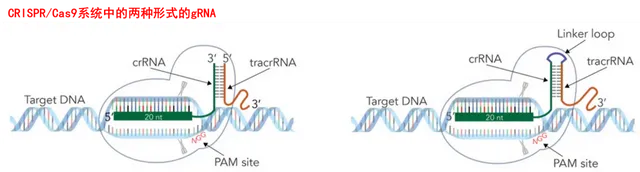
由成熟的crRNA與tracrRNA形成的gRNA是雙鏈RNA結構。實操中為了簡化操作,研究者將crRNA和tracrRNA使用linker連線,即構成了單精靈RNA(single guide RNA ,sgRNA),其作為精靈更簡便、更經濟。
4. BE
BE是基於CRISPR/Cas9技術開發的基因編輯工具,與上述基因編輯工具都是依賴於DNA雙鏈斷裂之後的HDR和NHEJ進行修復不同,BE提供了不引入DSB和外源DNA樣版條件下,就可以對單個堿基進行轉換的可能性。
BE目前主要有兩種,一種是胞嘧啶堿基編輯器(Cytosine Base Editors,CBEs),可以將C-G變成T-A,另一種是腺嘌呤堿基編輯器(Adenine Base Editors,ABEs),可以將A-T轉變成G-C。
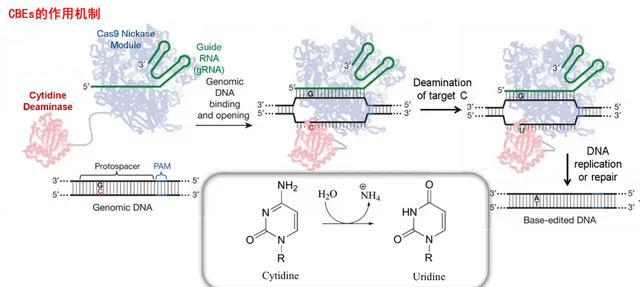
具體來說,BE系統主要由Cas9切口酶(Cas9n)、sgDNA和脫氨酶組成。BE使用的Cas9n是失活或部份失活處理的,但其可與目標DNA結合,使得sgDNA與目標序列的互補鏈結合,開啟DNA雙螺旋,此時由脫氨酶對靶位上的單個堿基進行轉變。而後,兩條DNA鏈堿基不配對,互補鏈經過DNA修復之後與新堿基配對,這樣就實作了單一堿基對的替換。
因此,BE系統這把「剪刀」的定位能力仍是由sgDNA提供的,修改能力則是由Cas9n和脫氨酶提供的。
註:Cas9的核酸酶剪下活性取決於兩個結構域:RuvC和HNH,它們分別負責切割DNA鏈的兩條鏈,且能單獨被人工點突變失活。突變包括RuvC催化結構域的第10位天門冬胺酸突變為丙胺酸(D10A)以及HNH催化結構域的第840位組胺酸突變為丙胺酸(H840A)。
前者發生則產生Cas9 D10A突變體,呈現RuvC失活(RuvC-)、HNH(HNH+)啟用的狀態;後者發生則產生Cas9 H840A突變體,呈現RuvC啟用(RuvC+) 、HNH失活(HNH-)的狀態。兩種突變體的Cas9仍然具有核酸酶活性,可對靶向序列進行單鏈剪下。
兩者同時發生,RuvC和HNH同時處於失活狀態時(D10A&H840A; RuvC-&HNH-),Cas9將不具有核酸酶活性,成為dCas9(dead Cas9)。dCas9雖然沒有剪下DNA的能力,但仍然可以在gRNA的引導下與特定的DNA序列結合。
對於CBEs系統,當Cas9n-胞嘧啶脫氨酶融合蛋白在sgRNA的引導下靶向基因組DNA時,胞嘧啶脫氨酶可結合到由Cas9n、sgRNA及基因組DNA形成的R-loop區的ssDNA(Single-Stranded DNA,單鏈DNA)處,將該處一定範圍內的C脫氨為U,進而透過DNA修復或復制將U轉變為T,最終實作C-T至G-A的轉換。
對於ABEs系統,當Cas9n-腺嘌呤脫氨酶 融合蛋白在sgRNA的引導下靶向基因組DNA時,腺嘌呤脫氨酶 可結合到由Cas9n、sgRNA及基因組DNA形成的R-loop區的ssDNA處,將該處一定範圍內的A脫氨為肌苷(I),肌苷在DNA水平會被當作G進行讀碼與復制,最終實作A-T至G-C的轉換。
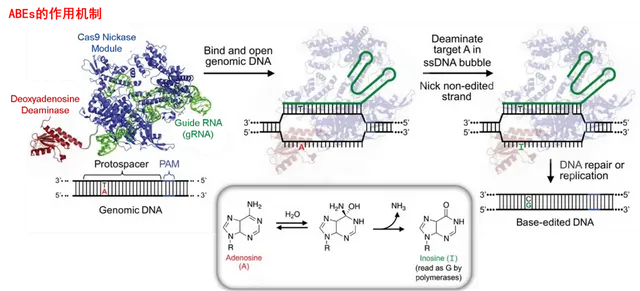
5. PE
PE是基於BE技術開發的基因編輯工具,簡單來說就是在BE技術的基礎上引入逆轉錄的方法,不僅可以實作單個堿基轉換,還可以實作堿基敲除、添加等多種操作。
PE系統主要由三個部份組成:Cas9 H840A切口酶、逆轉錄酶(Reverse transcripatse,又稱反轉錄酶,RT酶) ,以及起引導作用的pegRNA(prime editing guide RNA,PE精靈RNA)。
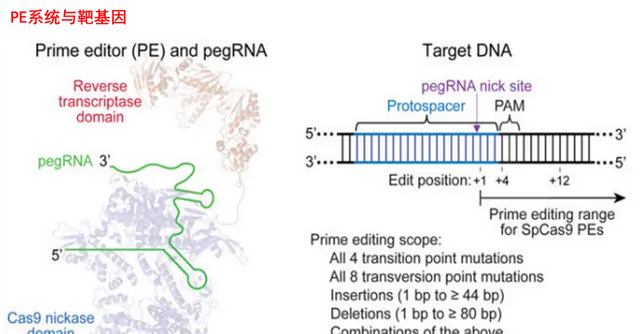
PE的作用機制是pegRNA的引導偶聯的Cas9 H840A和RT酶 靶向結合並切開一條DNA鏈,而後逆轉錄生成的單鏈DNA會在該切口處與原始序列展開競爭,由於DNA修復的酶一般從5端結合DNA鏈,所以原始的鏈傾向於被清除掉,希望加入的片段則被優先整合到原序列中。這樣即可實作堿基的替換、增減,以及DNA片段的增減。
因此,PE系統這把「剪刀」的定位能力是由pegDNA提供的,修改能力則是由偶聯的Cas9 H840A和RT酶 提供的。

三、不同基因編輯工具的比較
基因編輯工具的前進演化中,最重要的一點是為剪刀提供定位功能模組的變化。在ZFN之前,其實還有一種稱為兆核酸酶(Meganuclease)的基因編輯技術。
由於對DNA序列的辨識位點較大(>14 bp),兆核酸酶特異性高,且其自身編碼基因較小(僅1 Kb左右)易於傳遞。然而,兆核酸酶的DNA結合域和切割域不易區分,設計困難;一旦更換靶基因就要重新設計,做大規模蛋白質工程,成本較高。且多數兆核酸酶很難在人基因組上找到合適的位點,使其在一定程度上套用受限。
ZFN和TALEN把核酸酶的辨識域和切割域分開,因此換靶基因時只需客製辨識域就行(即只需做針對辨識域的蛋白質工程)。不同的是,TALEN蛋白結構具有重復胺基酸序列,相比ZFN鋅指結構域的設計挑戰較小,且靶向特異性更高,但TALEN在size上增大了遞送難度(~6kb vs.~2kb)。
到了CRIPSR/Cas系統,終於不需要折騰蛋白才能更改靶基因了,只需客製一個sgRNA就能靶向目標,設計難度、構建難度、綜合成本大大降低。此外,CRISPR/Cas的辨識精度、剪下效率也更高。
脫靶效應和細胞毒性方面,由於ZFN透過二聚體產生作用,一旦形成異源二聚體,就很可能造成脫靶效應,TALEN也一樣容易產生脫靶效應,只不過細胞毒性比ZFN要小。CRISPR/Cas的脫靶效應和細胞毒性都較低。
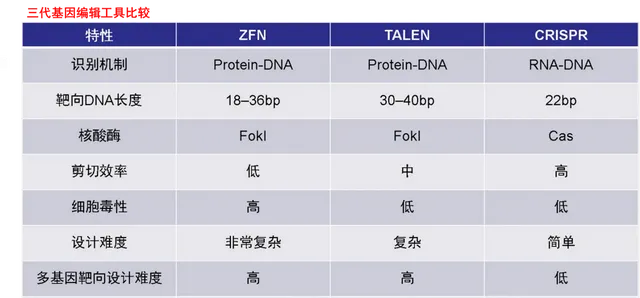
上述三代基因編輯工具的底層原理都是先引發DNA的DSB,再利用NHEJ或HDR對DSB進行修復。這會產生兩個問題。
一是兩種修復途徑中NHEJ總是占優勢的,很容易在基因敲入中引發基因剔除。因此在基因敲入中需要考慮如何增加HDR的概率(如CRISPR/Cas9各改進款式中,或者加NHEJ抑制劑從而減少NHEJ的占比,或者加HDR促進劑從而拉高HDR的占比)。此外,HDR發生在細胞分裂期,對於不分裂的細胞(如神經系統)很難實作基因敲入。
二是DSB可能是基因組中最嚴重的事故之一,這也是為什麽細胞會不惜錯誤也要用NHEJ緊急修復它。DSB帶來的風險包括基因組不穩定、片段改變、染色體易位以及致癌風險等。引發DSB意味著引入風險。
在這種情況下,不依賴DSB修復機制的基因編輯技術BE和PE應運而生。BE技術具有編輯效率高、編輯損傷少等特點,但是使用脫氨酶可能增加癌變的風險,而且可能出現的脫靶效應會嚴重影響編輯效率。
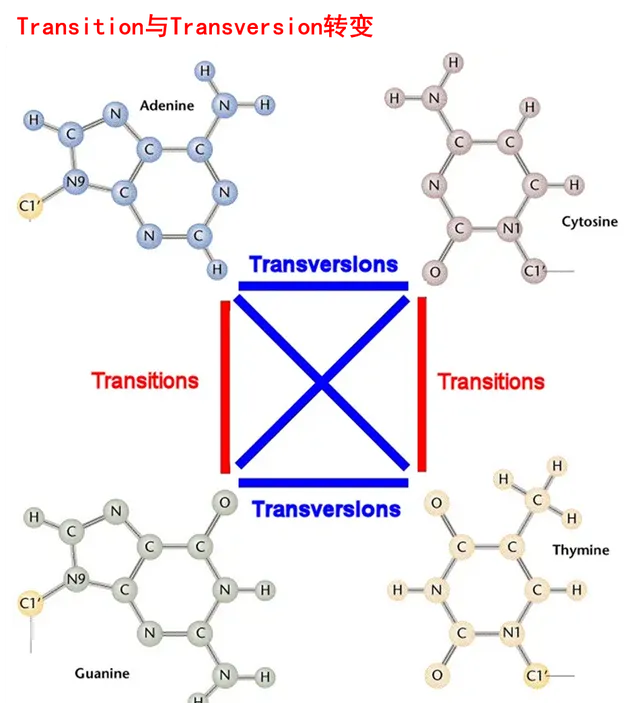
此外,BE只能做到嘌呤-嘌呤或者嘧啶-嘧啶的互相轉變(即transition),對嘌呤-嘧啶的互相轉變(即transversion)則無能為力。
PE則針對BE的脫靶現象進行了改進,不僅較好解決了脫靶的問題,而且可以同時實作transition和transversion,也就是說PE可以實作全部12中堿基的轉換。只不過PE技術還不成熟,其可靠性有待進一步研究,而且與BE技術相似,使用逆轉錄酶和脫氨酶仍然存在安全隱患。
至此,我們已經對基因編輯的原理、基因編輯工具及其對比有了清晰的了解。新技術的誕生與發展,不僅使其作為更精準、更高效的基因研究工具被開發,也因其在基因篩選、模型構建、機制研究中發揮了不可替代的獨特作用,為疾病治療提供了新思路、新範式,當然也為我們的偵察兵章魚托波的誕生提供巨大助力。
溫馨提示:本文僅是筆者思考內容的記錄,僅供讀者參考,不作為任何投資建議;投資有風險,入市需謹慎。











