孤兒G蛋白偶聯受體84(GPR84)是一種與癌癥、炎癥和纖維化疾病相關的受體。牛津大學報道了G蛋白受體偏向性激動劑 DL-175 ,其透過Gai/cAMP和β-arrestin不同訊號通路傳導,但代謝迅速。本文主要從 DL-175 開始進行分子最佳化,經SAR分析發現萘基引入取代基可以提高代謝穩定性,在吡啶N-氧化物基團上引入5-羥基得到化合物 68(OX04528) 和 69(OX04529) ,將cAMP訊號傳導的效力增強3個數量級,低至皮摩爾。兩個化合物在濃度最高達到80μM條件對β-arrestin招募沒有影響。化合物 68 和 69 是一種具有開發潛力的GPR84偏向性激動劑,可進一步評價體內藥效活性。本篇文章主要介紹了化合物 68 和 69 發現和分子最佳化過程,可為類似專案結構最佳化提供寶貴經驗。

圖1. 化合物68和69發現和分子最佳化過程
G蛋白偶聯受體(GPCR)是由人類基因組編碼的最大膜蛋白家族,調節多種不同的生理過程。GPCR超家族受體對多種配體都有反應。其中,遊離脂肪酸(FFAs)是一種必需營養素,對於心血管、新陳代謝和炎癥相關的眾多生理過程具有影響。以FFA為內源性配體調節其功能的GPCR家族視為遊離脂肪酸受體(FFARs),包括中長鏈FFARs, 如GPR40(FFA1)和GPR120(FFA4)以及短鏈FFARs,GPR43(FFA2)和GPR41(FFA3)。
G蛋白偶聯受體84(GPR84),視為rhodopsin類A GPCRs和假定的第五脂肪酸受體是其中之一。GPR84透過綜合表達序列標簽數據庫搜尋的方法發現的,然後從GPR84編碼的人外周血嗜中性球中複制和表征。GPR84主要表達於骨髓細胞,包括單核球、巨噬細胞、嗜中性球、嗜酸性球、佛波酯啟用的外周血單核球和位於中樞神經系統的小膠質細胞。鏈長為9-14飽和中鏈脂肪酸(MCFAs)是GPR84的激動劑,參與Gai訊號傳導,透過抑制腺苷酸環化酶來減少環磷酸腺苷(cAMP)的產生。
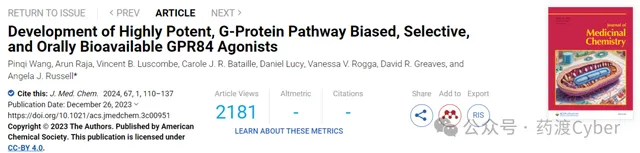
然而,最有效的MCFA——癸酸 1 (Fig1)以及MCFA氧化代謝產物,3-羥基十二烷酸 2 僅表現出微摩爾效力,無法招募β-arrestin,這與GPR84仍是孤兒受體的觀點一致。淋巴細胞和脂肪細胞中GPR84 mRNA表達可以透過維生素D、炎癥刺激物如LPS、腫瘤壞死因子α(TNFα)和慢性低度炎癥顯著上調。體外合成的激動劑啟用GPR84會導致吞噬作用增強、免疫細胞遷移和細胞因子、趨化因子和其他炎癥因子分泌增加。GPR84激動劑已報道可增強脂肪細胞質膜相關蛋白(APMAP)缺陷癌細胞的吞噬作用,抑制脂毒性誘導的巨噬細胞過度啟用,引發細菌粘附增加,顯示出抗動脈粥樣硬化作用,並在線粒體代謝調節中發揮作用,表明GPR84的啟用可能是有益於癌癥、殺菌和代謝功能障礙。
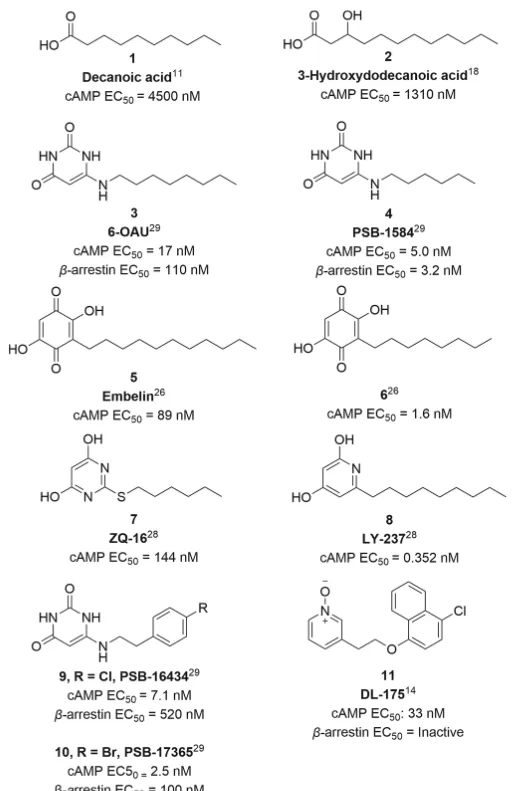
內源性的GPR84配體尚不清楚,6-辛基胺基尿嘧啶 3 ( 6-OAU )是第一個從化合物庫中篩選發現的合成激動劑。該化合物由一個極性投基團和親脂鏈組成的擬酸結構,是常用的陽性對照化合物,既能啟用G蛋白,又能招募β-arrestin。其衍生物 4 ( PSB-1584 )在G蛋白和β-arrestin途徑中顯示出增強的活性。 Embelin 5 (Fig1)是一種可以啟用GPR84的天然產物,發現其具有鎮痛、抗腫瘤、抗炎、抗氧化和傷口愈合活性。透過對脂肪鏈進行修飾,化合物 6 對GPR84的啟用表現出更高的效力和更好的選擇性。基於160,000種化合物的高通量篩選來自中國國家化合物庫,發現化合物 7 ( ZQ-16 )比化合物 6 更有效。經過SAR分析,發現化合物 8 ( LY-237 )在鈣離子和cAMP實驗檢測中在hGPR84轉染的CHO細胞中比化合物 7 和 3 更有效。
尿嘧啶衍生物 9 和 10 ( PSB-16434 和 PSB-17365 )報道對GPR84和G蛋白訊號傳導偏向具有增強的效力。最近有文獻報道了化合物 3 和 8 與GPR84結合冷凍電鏡結構,展現了GPR84啟用的蛋白結構基礎。在許多GPCR蛋白中,G蛋白和β-arrestin介導的訊號通路已被證明具有不同的生物醫學和生理作用,使得他們能夠將功能效應導向特定途徑。偏向激動劑可以辨識具有更高功效並減少脫靶副作用的化合物,開發偏向激動劑已成為一個日益活躍的研究領域。
牛津大學使用定量結構-活性關系(QSAR)模型進行虛擬篩選,進行初步的SAR分析得到了化合物 11 ( DL-175 )。在檢測cAMP積累抑制的實驗測定中,顯示出與化合物 3 相當的效力。然而,在測定最高濃度為60 μM下,它對β-arrestin招募沒有任何影響,表明對G蛋白訊號傳導存在顯著偏向。與化合物 3 相比,化合物 11 不能促進M1極化的U937巨噬細胞的趨化性,表明GPR84驅動的吞噬作用和趨化作用可以分開,並且誘導較少趨化性的偏向激動劑可能潛在的減少體內副作用。
此外,化合物 7 可以誘導GPR84在兩個蘇胺酸殘基(Thr263/Thr264)上的磷酸化,而化合物 11 沒有,且在GPR84受體中引入Arg172Ala突變不影響化合物 11 的活性,同時化合物 7 的活性喪失,這表明它們可能具有不同的結合模式。 然而,在小鼠肝細胞代謝實驗中,化合物11會快速代謝(t1/2<10mins),導致其不能作為體內工具化合物。 由於GPR84體外和體內偏向訊號傳導的影響仍有待認識和理解,因此需要開發具有適合體內研究的藥代動力學特征的偏向激動劑。牛津大學對化合物 11 進行分子最佳化成合適的體內工具化合物,用於臨床前模型中GPR84疾病生理學研究。經系統的SAR分析,最終開發出具有適當選擇性和ADME的GPR84有效、高度G蛋白訊號偏向的激動劑 68(OX04528) 和 69(OX04529) 的體內研究。
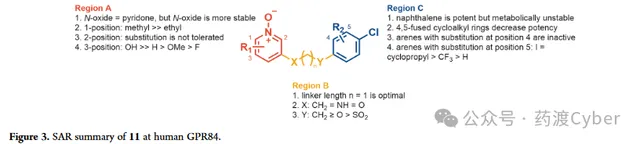
分子最佳化圍繞GPR84偏向激動劑 11 的化學反應以形成適合體內研究的分子改造策略,牛津大學首先試圖了解化合物 11 的代謝傾向。11與小鼠肝微粒體(MLM)和小鼠肝細胞一起孵育表現出快速代謝,t1/2分別為13.8mins和小於10mins。化合物 11 與小鼠肝細胞一起孵育60分鐘,並使用LC-MS/MS對所得代謝物進行表征:檢測到代謝物中有76%顯示單氧化,其中8%形成葡萄糖醛酸結合物;12%的代謝物是二羥基化,其余的12%是未鑒定的代謝物。鑒於代謝物情況,推測氧化主要發生在萘基團,因為萘酚和二氫二醇的氧化代謝較為常見。根據SAR分析以及對化合物11代謝物的認識,該結構被分為三個區域(Fig2),即疏水尾部(區域C)、連線基團(區域B)和極性頭基團(區域A)分別最佳化。
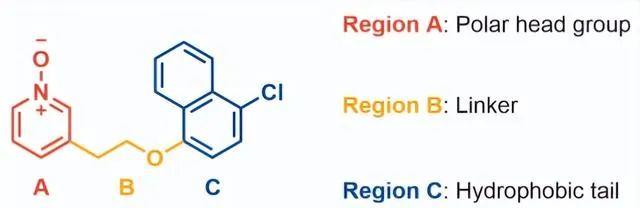
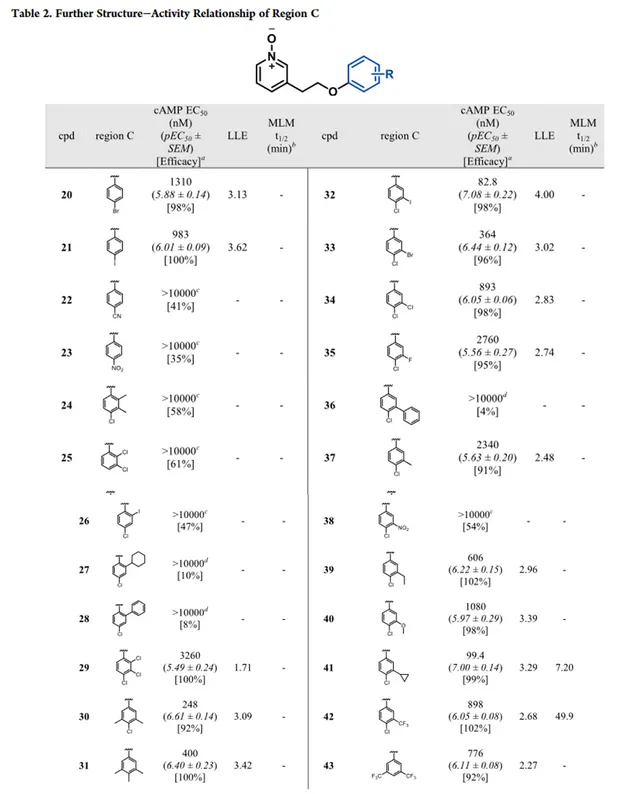
C區SAR分析
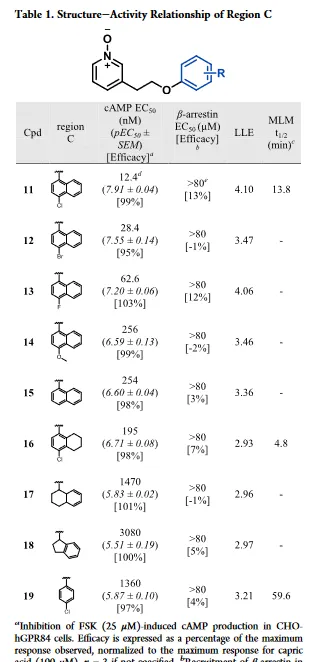
最初對化合物 11 的C區進行結構修飾,首先解決代謝不穩定的問題,如表1所示。透過抑制CHO-hGPR84細胞中毛喉素誘導的(FSK誘導)cAMP產生來測量GPR84激動劑的效力(EC50和pEC50±SEM)。透過最佳化分子的親脂性來提高化合物的體外活性,還檢測了親脂配體效率(LLE)。去掉4-氯取代基或用鹵素、給電子基團或氫替換4-氯取代結果說明了對鹵素的偏好。化合物 16 和 17 的效力降低15至118倍, 說明平面且為芳香基團是優選片段 。與化合物 17 相比,化合物 18 的效力降低,說明需要芳香基團。
對氯苯基取代的化合物 19 與 11 相比,其效力降低110倍。盡管與 11 相比,化合物 16 和 19 效力降低,但在代謝穩定性研究實驗發現,由於對氯苯基取代有利於代謝穩定性的提升( 19 ,MLM t1/2=59.6mins), 這與之前假設一致,即代謝主要位點是萘基 。由於效力降低且不存在其它親水基團,結構改造的化合物沒有高於化合物 11 的LLE。所有活性化合物與陽性對照物(癸酸)在cAMP實驗中相比都是完全激動劑。全部化合物在β-arrestin實驗中都沒有活性(如表1),這說明 11 的衍生物在GPR84上具有一致的G蛋白訊號偏倚。 根據化合物19的實驗結果,進一步探索C去取代芳烴以維持代謝穩定性並增強效力。
進一步研究C區的苯基取代,設計並合成了對位取代衍生物(表2)。對位鹵代衍生物 20 和 21 的效力與化合物 19 相當,與萘系列一致。對位取代的化合物 22 和 23 表現出效力顯著降低,可能是由於氰基和硝基降低了芳烴電子雲密度不利於氫鍵受體形成,或降低了疏水性。鄰位取代的衍生物 24-29 效力降低或減弱。與化合物 25 和 31 相比,化合物 29 和 30 效力增加進一步強調了鹵素作為對位取代基團的重要性。
間位碘取代的化合物 32 在LLE增強的情況下,效力提高20倍,說明適當的間位取代衍生物的有可能增強效力。不同的間位取代衍生物( 33-40 )效力降低進一步了解空間效應和活性之間的關系。將鹵素從碘取代的 32 改為空間阻礙較少的溴 33 、氯 34 和氟 35 ,活性逐漸降低。隨著受阻的3-苯基取代36的效力顯著降低。而較小的3-甲基、3-甲氧基和3-乙基化合物活性表現為乙基>甲氧基>甲基。間位硝基取代的 38 沒有表現出活性,而3-環丙基取代的41效力顯著提升,但相比於 32 ,LLE略有下降,表明間位取代需要適當平衡空間位阻效應和分子親脂性。
然而,在MLM研究中,發現化合物41快速代謝(MLM t1/2=7.2min),這可能是由於環丙基先被氧化所致。4-Cl-3-CF3取代的 42 和3,5-diCF3-取代的 43 與化合物 19 相比顯示出效力增強,且化合物42表現出良好的代謝穩定性(MLM t1/2=49.9min)。與陽性對照癸酸相比,cAMP實驗中發現所有活性化合物都是完全激動劑,並且在招募β-arrestin沒有檢測到活性,這與化合物 11 的結果一致。
B區SAR分析
接下來牛津大學研究連線子部份(B區),以探討鏈長、雜原子的引入/去除以及所選官能團的耐受性的影響(表3)。
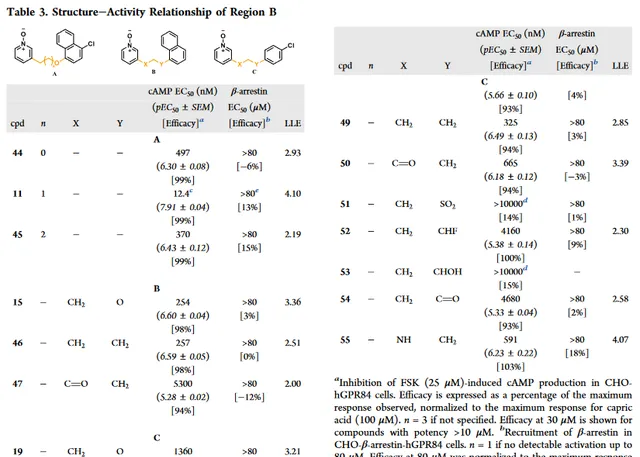
從化合物 44 、 45 和 11 相比較,很明顯,連線子的長度在3個原子時是最佳的。化合物 46 和 11 比較,說明另一個連線子不是必需的且可以用烷基連線子代替。但是,換成烷基連線子可能會降低LLE。與化合物 15 和 46 相比, 47 的效力下降20倍,表明引入羰基削弱效力。與化合物 19 相比,由於不同位置的醚鍵連線子 48 導致效力下降2倍。有趣的是,帶烷基連線子 49 和 19 相比,效力增加4倍。這與萘系列 46 不一致但顯示出具有保持效力和LLE的同時替代萘的潛力。
進一步替換 49 中X位為羰基得到化合物 50 ,效力降低2倍。透過改變對氯苯基衍生物上的位置(Fig3C),如碸 51 和羥基 53 效力減弱。與化合物 19 相比,氟和羰基使效力降低10倍以上。在化合物 55 的X位置引入胺基導致類似的效力,LLE高於化合物 19 ,但效力低於 49 。與陽性對照癸酸相比,cAMP實驗測定所有活性化合物都是全激動劑,並且在招募β-arrestin沒有檢測到活性。
N-氧化物吡啶的生物電子等排
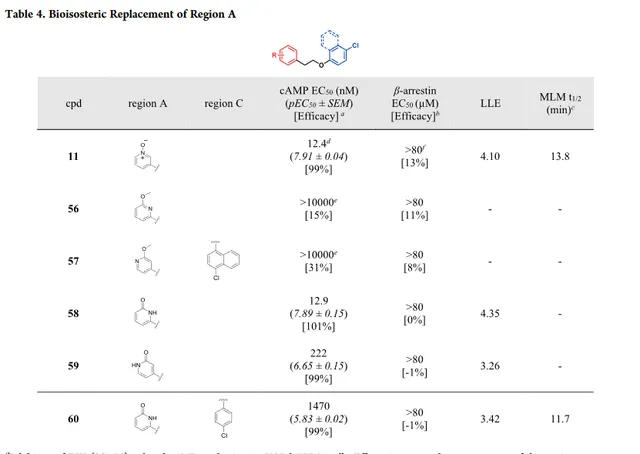
開始探索A區SAR研究之前,首先探索了非等排體替代策略,透過醛氧化酶代謝來潛在地減少吡啶N-氧化物分解。由於結構相似,氫鍵受體相似,吡啶酮可以是吡啶N-氧化物的生物等排體。O-甲基保護的羥基吡啶 56 和 57 在cAMP實驗測定中沒有活性,可能是由於甲基取代阻斷氫鍵受體作用(表4)。取代的吡啶酮 58 在cAMP實驗測定中顯示完全激動活性,β-arrestin效力和LLE與化合物 11 相當,說明吡啶酮與吡啶N-氧化物具有類似的作用。4-取代吡啶酮 59 的效力降低了17倍,可能表明氫鍵供體N-H減弱效力。因此,將化合物 60 作為化合物 19 的生物等排體,比較代謝穩定性。 然而,與吡啶N-氧化物19相比,化合物60(MLM t1/2=11.7min)穩定性較差,說明吡啶酮部份是體內代謝穩定性差的主要來源。因此,沒有進一步研究吡啶酮,開始研究吡啶N-氧化物基團修飾作為替代方案。
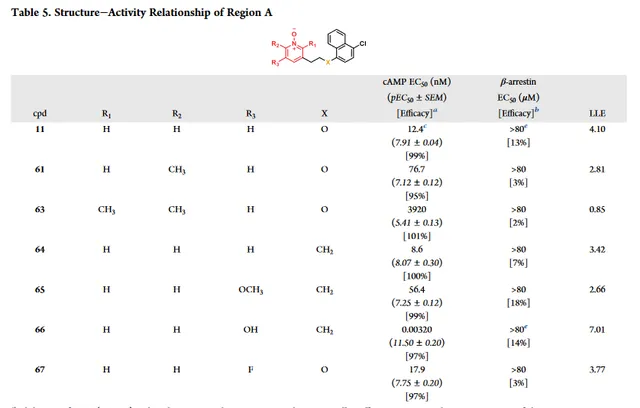
A區SAR分析
A區研究重點是透過改變R1、R2和R3的取代來增強活性(表5)。用甲基或乙基( 61 和 62 )取代R2表現出中等至大幅的效力下降,說明該位置存在空間限制。相比化合 61 ,化合物 63 在R1引入甲基活性顯著降低,表明R1的取代是不可接受的。透過將醚連結子轉為烷基連線子,與化合物 11 相比,化合物 64 顯示出相當的效力,且LLE略有下降,這與Fig3的萘系列一致。然而,透過抑制cAMP水平,在R3引入羥基作為取代基,效力增加2000倍。與陽性對照分子癸酸相比,起到了黃曲黴素的作用,同時,在最高測試濃度(80μM)條件下,仍沒有表現出β-arrestin募集作用。註意到化合物 66 的A區與 6-OAU 和 PSB-16434 內的嘧啶二酮有一定的結構相似性,使得牛津大學推測額外的羥基是否可以起到氫鍵受體作用,類似於 6-OAU 和 PSB-16434 的嘧啶二酮互變異構形式與GRP84的結合模式。
然而,最重要的是,在R3( 67 和 65 )導致活性降低,表明R3增加新的氫鍵供體實際上可能是負責增強效力。這些實驗觀察結果可能表明化合物 66 分別與 ZQ-16 和 LY-237 的二羥基嘧啶和二羥基吡啶互變異構體更接近。化合物 66 的這種增強效力可能是由於與GPR84中的相鄰殘基(與Arg172)形成新的氫鍵,因為化合物 11 透過獨立於Arg172的機制啟動GPR84啟用,對於其它類似脂質配體至關重要。 隨著效力增強,化合物66的LLE增加到超過5,完全再藥物發現專案的可接受範圍內。
SAR總結
化合物 11 周圍不同的3個區域(Fig3):區域A不能耐受位置1和位置2的取代,而位置3引入氫鍵供體使效力提高≥1000倍,發現3個原子接頭長度對於B區是最佳的;C區是主要的代謝位點,但透過用芳烴取代萘基團可以緩解。這導致活性喪失,可以透過位置5(Fig3,C區)上引入取代基並將Y更改為Cp(Fig3,B區)來緩解。
高效、有偏向且代謝穩定的激動劑設計
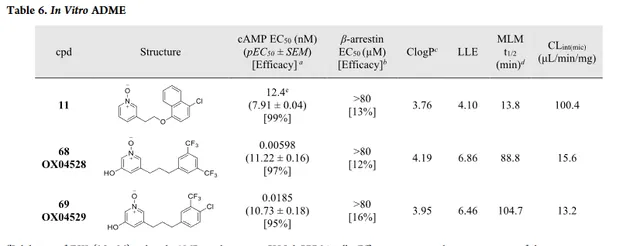
化合物 68 ( OX04528 )和 69 ( OX04529 )是透過將三個區域的主要發現合並在一起設計和合成的(表6)。
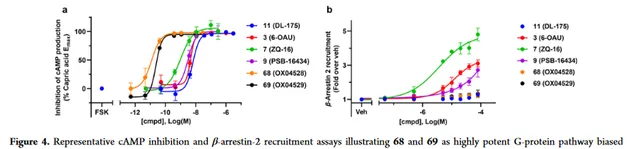
化合物 68 和 69 均表現出高效力和G蛋白訊號傳導偏差。一般認為,受體表達水平會影響基於細胞的測定中表觀效力的測量,較高的GPR84表達水平為同一配體提供較高的表觀效力值。為了避免這一潛在的混淆問題,牛津大學比較了化合物 68 和 69 在cAMP和β-arrestin招募測定的影響,以及偏倚程度不同的參考配體,比較相對效價值,其中包括 6-OAU 、 ZQ-16 和 PSB-16434 。在cAMP實驗測定中,效價排序為 68>69>ZQ-16>PSB-16434>6-OAU>11 。
在β-arrestin測定中, ZQ-16>6-OAU> PSB-16434>11=68=69 。與此前報道的配體相比,化合物 68 和 69 在cAMP抑制中表現出極大的改善活性(Fig4a和表2),而兩種配體在測試最高濃度下均為表現出β-arrestin募集(Fig4b)。這可能是由於與其他配體相比的不同結合模式,與之前觀察到 6-OAU 和 DL175 之間藥理學差異一致。這些差異需要用化合物 68 和 69 進行進一步研究。
體外ADME分析顯示化合物 68 和 69 在MLM中緩慢降解(t1/2=89min和105min)。比化合物 42 和 19 代謝更穩定,表明3位羥基也可能減慢吡啶N-氧化物的代謝。隨著效力增加3個對數級,化合物 68 和 69 均顯示出改善的LLE>5。
新GPR84激動劑的選擇性
為了確認化合物 68 和 69 誘導的cAMP產生的抑制是由GPR84介導的,對未轉染的CHO-K1親代細胞進行cAMP檢測,未檢測到cAMP的抑制(Fig5a)。根據先前發表的藥理學證據,化合物 11 的結合被認為與 6-OAU 和 MCFA 的結合位點重疊,盡管可能具有不同的結合模式,因為受體突變研究表明GPR84 Arg172 Ala突變消除了 MCFA 和 6-OAU 的相互作用,但化合物 11 沒有。化合物 11 在β-arrestin 募集激動劑和拮抗劑篩選試驗中顯示出 GPR84對168個人類GPCR的高選擇性。 MCFA 啟用GPR84表明 68 和 69 對脂質傳感器FFA1和FFA4可能存在潛在的脫靶效應。感應大麻素受體2(CB2)可被癸酸1啟用在微摩爾範圍內,因此對這三個 GPCR 進行了選擇性反篩選測試。用熒光成像酶標儀 (FLIPR) Ca2+ 測定(Fig5b-d) 68 和 69 ,顯示 FFA1、FFA4和CB2無活性。
體外細胞毒性
毒性是藥物開發中必須考慮的因素。因此,透過測量CHO-hGPR84細胞和CHO-K1細胞的乳酸去氫酶(LDH)釋放來檢查化合物 68 和 69 的細胞毒性。在所有測試濃度(高達30μM)下孵育20小時後,化合物 68 和 69 均未顯示出細胞毒性的證據。
體內藥代動力學研究
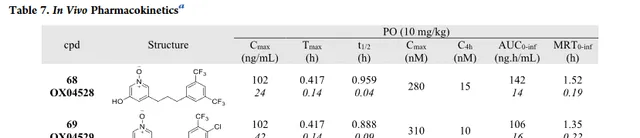
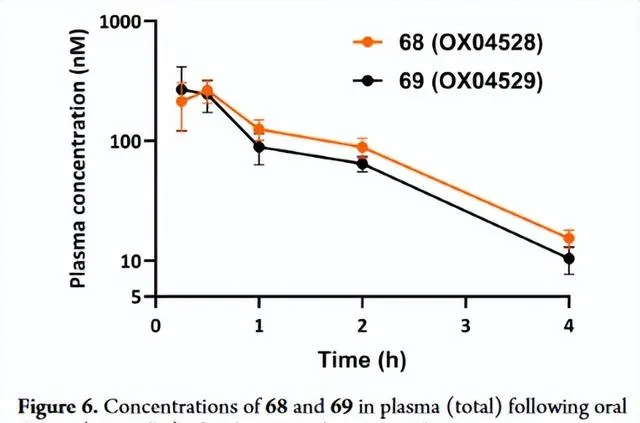
由於化合物 68 和 69 在體外測定中都表現出顯著增強的效力、訊號偏倚和改善的代謝穩定性,因此兩者都被用於體內PK分析。兩種化合物均具有口服生物利用度,並且分別具有58和53分鐘的適當體內半衰期(表7)。以10mg/kg口服給藥後,4小時後,兩種化合物在血漿中的總濃度約為10nM,遠高於細胞效力的測定值,支持它們進入體內功效研究(Fig6)。總而言之,這些數據表明這兩種化合物 68 和 69 將是有用的體內探針,值得進一步研究。
結論
強效GPR84偏向激動劑 68 和 69 的發現始於用4-氯苯基替代 11 中的對氯萘(C區,Fig2)導致效力降低100倍但代謝穩定性增強。區域的進一步最佳化導致了活性的一些恢復並保持了MLM的穩定性。嘗試用吡啶酮生物等排替代N-氧化物未能改善活性或代謝穩定性。對A區的SAR研究發現,與化合物 11 相比,添加α羥基可導致3個對數的效力增強,同時保留較高的訊號偏差,這透過其在β-arrestin招募測定中的無活性來證明。為了開發具有合適的藥物代謝和藥代動力學特征的候選藥物,設計並合成了化合物 68 和 69 。兩種工具化合物都顯示出極高的效力,EC50值分別為5.98和18.5pM,並且在β-arrestin測定中未檢測到活性(Fig4)。
此外,這些化合物對FFA1、FFA4和CB2受體具有選擇性。細胞暴露於 68 和 69 達到20小時並沒有導致LDH釋放,表明這些化合物在體外維持了細胞活力和低細胞毒性。化合物 68 和 69 的體外代謝研究顯示MLM穩定性增強。在小鼠PK研究中,化合物 68 和 69 的口服給藥顯示體內半衰期分別為58和53分鐘。然而,鑒於這兩種化合物的高活性,血漿中的總濃度在4h時高於各自的EC50,這為進一步的體內研究提供了機會。
參考來源
J. Med. Chem. 2024, 67, 110−137
來源:藥渡Cyber












