
马克思·普朗克煤炭研究所Benjamin List教授和北海道大学Nobuya Tsuji教授等在Science上报道题为「Catalytic asymmetric fragmentation of cyclopropanes」的文章。
作者介绍

德国马克斯普朗克煤炭研究所所长 本杰明·李斯特(Benjamin List) 教授与美国科学家麦克米伦一起荣获了2021年诺贝尔化学奖。本杰明·李斯特,1968年1月出生于德国法兰克福一个富裕的家庭。科学研究的基因或许早就植根于这个家庭中。李斯特的曾曾祖父雅各布·福尔哈德(1834-1910) 是著名的化学家,发现了Volhard–Erdmann环化反应。曾祖父弗朗茨·沃尔哈德以肾病学家的身份而闻名。
本杰明·李斯特教授的研究兴趣集中于有机催化与合成,他是不对称有机催化领域的开创者之一,发展了一种新型不对称催化模式:手性抗衡阴离子导向的不对称催化(ACDC)。这意味着在无金属小分子的帮助下加速化学反应的过程,并将它们选择性地引导到某些方向。这种方法近年来迅速发展,已经发挥着重要作用,例如使药物的制造更加环保。而以这种方式获得的知识也提供了关于某些对生命起源很重要的分子最初是如何形成的线索。
烷烃的立体选择性转化一直是化学领域的关键挑战。虽然目前金属酶催化剂能够对烷烃的氧化具有优异的表现和选择性,但是烷烃的化学转化方法一直局限于过渡金属催化C-H键官能团化。在强Brønsted酸作用下,烷烃能够质子化生成5配位的碳正离子,随后分解为更小的烷烃化合物。但是如何控制这个过程进行的立体选择性仍然没有方法。有鉴于此, Benjamin List教授和Nobuya Tsuji教授 报道发现限域的强酸能够以立体选择性的方式将许多环丙烷化合物切断,转化为烯烃化合物。这个方法拓展了烷烃活化的领域,通过理论计算说明这个反应过程有可能包括环丙基阳离子(一种非典型结构碳正离子)。
反应设计和发展
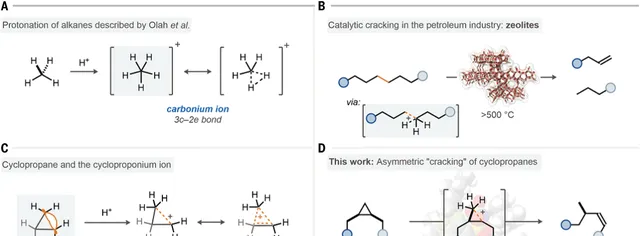
图1. 反应的研究背景
首先研究双环[3,1,0]己烷( 1a )的3-取代反应。作者设计分子不对称加氢的反应进行去对称化,随后脱质子和异构反应生成烯烃。当使用比较弱的Brønsted酸作为催化剂(磷酸、IDP(亚胺二磷酸盐,imidodiphosphoric acid)、iIDP(空间限域亚胺二膦酸盐)),只能得到非常差的环丙烷反应活性。但是,亚氨基二磷酰亚胺酯IPDi(imidodiphosphorimidates) 7a 具有良好的反应性,能够生成 2a - 6a 烯烃混合物。其中主要的产物是三个取代基修饰的烯烃 2a 和 3a ,产物主要为消旋形式,另外含有少量其他产物( 4a, 5a, 6a )。
催化剂的设计与优化。作者发现除了立体位阻作用,增强催化剂和反应物之间的非共价相互作用对控制反应的位点选择性和立体选择性非常重要。作者提出BINOL(1,1′-双-2-萘醇)的3,3′位点取代基能够形成催化微环境,氮原子上的取代基决定了阴离子的碱性,实现了控制与反应物之间的静电相互作用。因此,IDPi催化剂 7b,(BINOL取代基-叔丁基结构,增强立体位阻和静电相互作用),对生成2a-5a 具有更高的区域选择性和立体选择性。通过将催化位点的三氟甲磺酰基替换为芳基磺酰基,增强催化位点的碱性,并且通过π-π堆叠作用提高构象的刚性,催化剂IDPi 7c 得到更好的位点选择性和立体选择性。
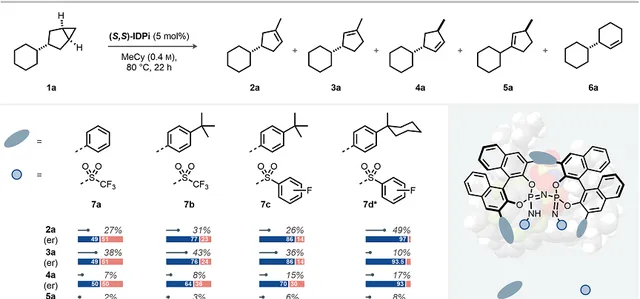
图2. 催化剂和反应条件优化
反应效果。之后再次对催化剂和反应条件进行优化,发现IDPi 7d 具有最好的催化反应效果,得到令人满意的产率和立体选择性,优化了反应温度和反应时间之后,能够以40 %的产率和97:3的立体选择性生成环戊烯产物 2a 。另外还有少量 3a , 4a , 5a 生成,产率分别为6%,13%,7%,而且都具有优异的立体选择性。此外发现适中的立体选择性生成少量 6a 。
反应兼容
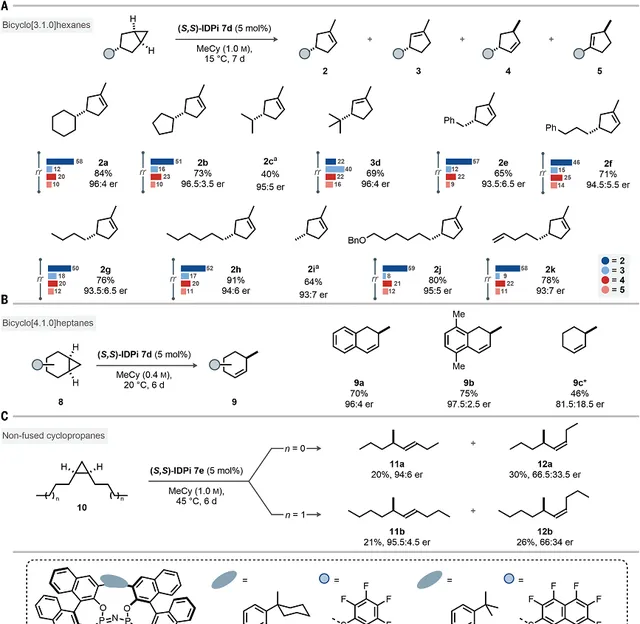
图3. 反应物兼容
研究反应的兼容性。
二级烷基取代基。反应对环烷( 2a 和 2b )和非环烷( 2c )二级烷基取代基兼容,另外对立体拥挤的三级取代基兼容,能够以优异产率和立体选择性生成 3d 。
一级烷基取代基。反应对一系列一级烷基取代基修饰的反应物,包括苄基( 2e )、3-苯基丙基( 2f )、丁基( 2g )、己基( 2h )都能够兼容并且获得优异的立体选择性。生成的烯烃 3e 能够通过氧化反应生成醛。令人意外的是,对于较小的甲基取代基,反应仍能够以64%的收率和93:7的立体选择性生成对应的 2i 产物。 这个结果说明IDPi催化剂对于控制尺寸较小的简单结构烷烃的优异效果。 反应对于苄氧基( 2j )或者端基烯烃( 2k )取代基的烷烃同样得到优异的产率和立体选择性,说明该反应的优异官能团耐受性。生成的产物能够通过加氢、Ritter反应、傅-克烷基化反应、甲基化-加氢反应生成其他功能型化合物。这些转化过程不会消耗分子的立体选择性。这个反应能够用于双环[4,1,0]庚烷( 8 )的转化。由于催化剂对苄基碳正离子具有稳定作用,因此对于苄氧基环己烯( 9a 和 9b )能够获得高产率和优异的立体选择性。对于结构特别简单的双环[4,1,0]庚烷,能够以有价值的产率和立体选择性生成环己烯 9c 。
该反应甚至能够对非稠环化的环丙烷化合物( 10 )进行立体选择性转化,该反应化合物具有的柔性是导致其转化面临挑战性的原因。这个反应方法能够在更高的温度下进行1,2-双丙烷-环丙烷( 10a )的转化,以适中的产量生成双官能团取代的手性烯烃分子( 11a , 12a )。该反应同样兼容1,2-双丁基环丙烷( 10b )。
机理研究
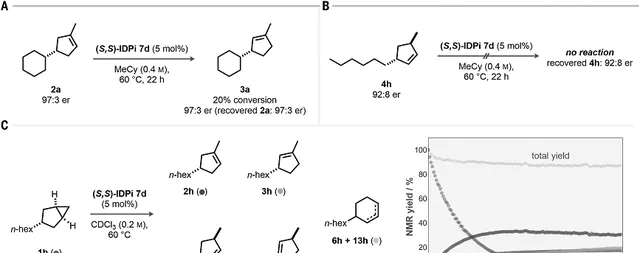
图4. 反应机理研究
控制实验。反应机理的一个问题是反应立体选择性产生的原因来自「不对称质子化」还是「产物的动力学拆分」。对 2a 在60℃使用催化剂 7d 处理,发现20%的 2a 异构化生成 3a,但是2a 和 3a 的立体结构比例并没有改变。而且,反应生成的 3a 的对映体比例低于 2a ,说明 2a 的异构不可能是生成 3a 的主要步骤。通过烯烃4h的反应情况同样验证催化反应不是异构化过程。通过时间分辨 1 H NMR研究,发现在60℃进行反应,1天内产物 4h , 5h , 6h , 13h 达到最大值并且不再增加,但是 3h 烯烃仍不断增加, 2h 不断消耗。
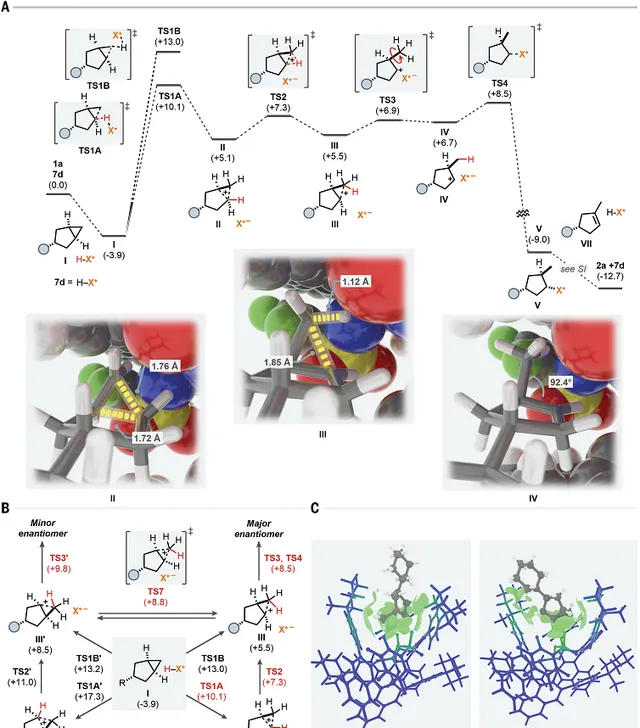
图5. 理论计算机理研究
DFT理论计算。通过DFT理论计算研究 7d 催化剂和 1a 的反应路线和反应机理。首先,反应物进入IDPi催化剂的内部,形成 I 复合物。随后IDPi催化剂对催化剂内的反应物质子化。质子化生成碳正离子 II 或者非典型的碳正离子 III ,通过立体选择性的质子化,分别还可能生成 II′ 或 III′ 。而且中间体 II 或 II′ 能够通过1,3-H原子转移生成 III 或 III′ 。但是步骤具有不同的能垒。生成 III 中间体之后,甲基旋转( TS3 )导致产生二级碳正离子 IV ,随后环翻转与IDPi催化剂自发形成共价键的加合物 V ;生成 III′ 中间体后,甲基旋转,随后通过 TS3′ 生成加合物 V′ ,生成次要立体结构的产物。能量计算发现 TS4 和 TS3′ 之间的能量差为1.3kcal/mol,这个计算的结果与实验结果符合(1.7kcal/mol, 298.15K)。反应中生成的其他产物可能通过中间体的重排和脱质子化过程生成。
非共价相互作用的作用。通过IGMH方法研究非共价相互作用对立体选择性的作用(Multiwfn软件,一款国产软件)。发现反应物能够很好的稳定在 7d 催化剂的 TS4 过渡态,同时取代基在口袋外部。但是, TS3′ 过渡态导致口袋被反应物撑开。计算结果明确展示了反应物和BINOL之间的强vdW相互作用。对质子化步骤 TS1A 、 TS1B 、 TS1A′ 、 TS1B′ 进行计算,发现催化剂的变形和相互作用对于控制反应选择性非常重要。
参考文献及原文链接
Ravindra Krushnaji Raut, Satoshi Matsutani, Fuxing Shi, Shuta Kataoka, Margareta Poje, Benjamin Mitschke, Satoshi Maeda, Nobuya Tsuji*, Benjamin List*, Catalytic asymmetric fragmentation of cyclopropanes, Science 386, 225–230 (2024)
DOI: 10.1126/science.adp9061
https://www.science.org/doi/10.1126/science.adp9061











