神经系统的线粒体转运机制是一个复杂而重要的过程,涉及多个细胞结构和信号通路。以下是一个详细的描述,并附有相关的参考文献。
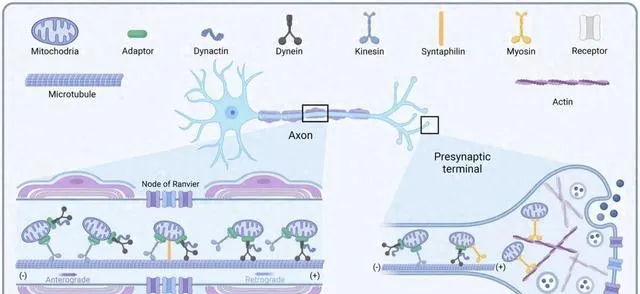
神经系统中线粒体的转运机制主要包括以下几个方面:
线粒体动态平衡
神经元中的线粒体需要在细胞内进行动态平衡,包括在轴突、树突和细胞体之间的双向运输。这种平衡有利于满足不同区域的能量需求,同时也有助于维持细胞内的Ca2+稳态,从而调节神经递质的释放和信号传递 [1]。线粒体的双向运输依赖于细胞骨架,主要通过微管上的动力蛋白进行。kinesin家族的蛋白负责线粒体向轴突末端的运输,而dynein家族的蛋白则负责向细胞体的逆向运输 [2]。这些动力蛋白通过与线粒体外膜上的受体蛋白(如Miro、Milton等)结合来实现线粒体的定向运输。
线粒体融合和分裂
线粒体的运输过程中,还需要发生融合和分裂,以调节其形态和功能。线粒体的融合主要由OPA1(optineurin atrophy 1)、Mfn1/2(mitofusin 1/2)等蛋白介导,这些蛋白位于线粒体外膜和内膜上,通过相互作用促进膜融合。而线粒体的分裂则主要由Drp1(dynamin-related protein 1)介导,Drp1可以结合到线粒体外膜上并引起膜的收缩和分裂 [3]。线粒体的融合有利于维持完整的功能,而分裂则可以产生独立的线粒体单元以满足局部的能量需求。这种动态平衡对神经元的能量代谢和信号传递至关重要。
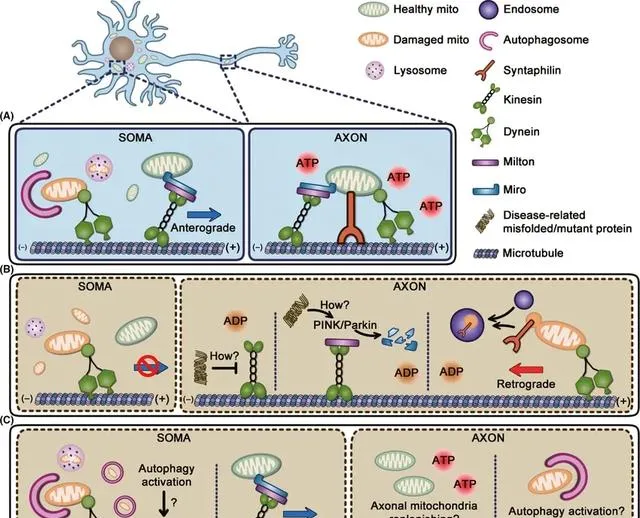
胞质Ca2+对线粒体的调控
Ca2+作为一种重要的细胞信号分子,在神经系统中对线粒体的转运和功能也有重要调控作用。当神经元兴奋时,细胞内Ca2+浓度会上升。这些Ca2+可以通过Miro蛋白结合到线粒体表面,促进kinesin和dynein蛋白与线粒体的结合,从而调节线粒体在神经元内的双向运输 [4]。同时,胞质Ca2+的升高也会促进线粒体的融合和分裂,从而调整其在细胞内的分布。此外,线粒体内膜上的Ca2+uniporter可以将细胞质的Ca2+吸收进入线粒体基质,参与调节细胞的能量代谢和信号传递。这种Ca2+的双向转运在神经元的兴奋-抑制平衡中扮演重要角色 [5]。
神经营养因子的调控
神经营养因子如NGF(神经生长因子)、BDNF(脑源性神经营养因子)等,也可以调节神经元中线粒体的转运和功能。这些神经营养因子可以通过激活特定的受体(如TrkA、TrkB),进而激活下游的信号通路,如PI3K-Akt、MAPK等。这些信号通路可以增强kinesin和dynein蛋白与线粒体的结合,促进线粒体在神经元内的双向运输 [6]。同时,神经营养因子还可以调节线粒体的融合与分裂,从而影响其形态和功能。例如BDNF可以抑制Drp1的活性,减少线粒体的分裂,维持其完整性 [7]。
病理条件下的线粒体异常
在一些神经系统疾病中,线粒体的转运和功能受到严重损害,这些都会影响神经元的能量供给和细胞信号传递,最终导致神经细胞的损伤和死亡。例如在阿尔茨海默病中,tau蛋白的异常磷酸化会破坏微管结构,影响kinesin和dynein蛋白与线粒体的结合,从而减弱线粒体在神经元内的双向运输 [8]。在帕金森病中,PINK1和Parkin介导的线粒体清除(mitophagy)过程受损,使得受损的线粒体无法被及时清除,导致线粒体功能障碍和神经细胞死亡 [9]。此外,在运动神经元疾病中,SOD1基因突变也会影响线粒体的动态平衡,加剧神经细胞的损伤 [10]。
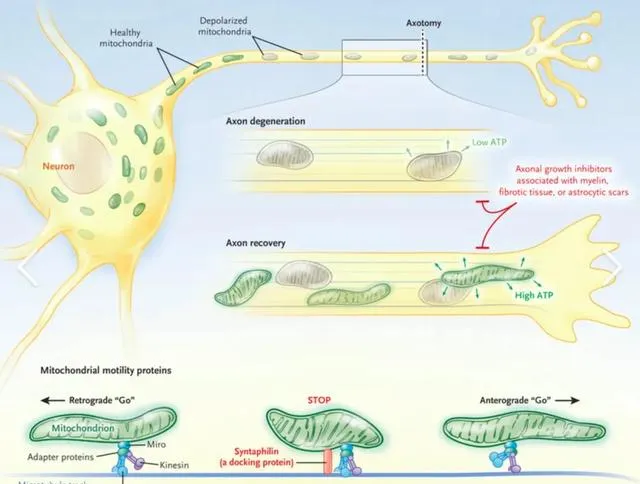
综上所述,神经系统中线粒体的转运机制涉及多个层面,包括线粒体在神经元内的动态平衡、融合分裂,以及各种细胞内信号的调控。这些过程对维持神经元的能量代谢和信号传递至关重要。而在一些神经系统疾病中,线粒体的异常转运和功能障碍都会导致神经细胞的损伤。深入了解这些机制有助于更好地认识神经系统的生理特点,并为相关疾病的预防和治疗提供新的思路。
在一些神经系统疾病中,线粒体的转运和功能受到严重损害,这些都会影响神经元的能量供给和细胞信号传递,最终导致神经细胞的损伤和死亡。例如在阿尔茨海默病中,tau蛋白的异常磷酸化会破坏微管结构,影响kinesin和dynein蛋白与线粒体的结合,从而减弱线粒体在神经元内的双向运输 [8]。在帕金森病中,PINK1和Parkin介导的线粒体清除(mitophagy)过程受损,使得受损的线粒体无法被及时清除,导致线粒体功能障碍和神经细胞死亡 [9]。此外,在运动神经元疾病中,SOD1基因突变也会影响线粒体的动态平衡,加剧神经细胞的损伤 [10]。神经系统中线粒体的转运机制涉及多个层面,包括线粒体在神经元内的动态平衡、融合分裂,以及各种细胞内信号的调控。这些过程对维持神经元的能量代谢和信号传递至关重要。而在一些神经系统疾病中,线粒体的异常转运和功能障碍都会导致神经细胞的损伤。深入了解这些机制有助于更好地认识神经系统的生理特点,并为相关疾病的预防和治疗提供新的思路。
参考文献
[1] Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13(2):77-93.
[2] Saxton WM, Hollenbeck PJ. The axonal transport of mitochondria. J Cell Sci. 2012;125(Pt 9):2095-2104.
[3] Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79-99.
[4] Macaskill AF, Rinholm JE, Twelvetrees AE, et al. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541-555.
[5] Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda). 2008;23:84-94.
[6] Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14(14):1272-1276.
[7] Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16(6):899-909.
[8] Kandimalla R, Reddy PH. Multiple Faces of Dynamin-Related Protein 1 and Its Role in Alzheimer's Disease Pathogenesis. Biochim Biophys Acta Mol Basis Dis. 2016;1862(4):814-828.
[9] Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119-131.
[10] Magrane J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11(7):1615-1626.











