孤儿G蛋白偶联受体84(GPR84)是一种与癌症、炎症和纤维化疾病相关的受体。牛津大学报道了G蛋白受体偏向性激动剂 DL-175 ,其通过Gai/cAMP和β-arrestin不同信号通路传导,但代谢迅速。本文主要从 DL-175 开始进行分子优化,经SAR分析发现萘基引入取代基可以提高代谢稳定性,在吡啶N-氧化物基团上引入5-羟基得到化合物 68(OX04528) 和 69(OX04529) ,将cAMP信号传导的效力增强3个数量级,低至皮摩尔。两个化合物在浓度最高达到80μM条件对β-arrestin招募没有影响。化合物 68 和 69 是一种具有开发潜力的GPR84偏向性激动剂,可进一步评价体内药效活性。本篇文章主要介绍了化合物 68 和 69 发现和分子优化过程,可为类似项目结构优化提供宝贵经验。

图1. 化合物68和69发现和分子优化过程
G蛋白偶联受体(GPCR)是由人类基因组编码的最大膜蛋白家族,调节多种不同的生理过程。GPCR超家族受体对多种配体都有反应。其中,游离脂肪酸(FFAs)是一种必需营养素,对于心血管、新陈代谢和炎症相关的众多生理过程具有影响。以FFA为内源性配体调节其功能的GPCR家族视为游离脂肪酸受体(FFARs),包括中长链FFARs, 如GPR40(FFA1)和GPR120(FFA4)以及短链FFARs,GPR43(FFA2)和GPR41(FFA3)。
G蛋白偶联受体84(GPR84),视为rhodopsin类A GPCRs和假定的第五脂肪酸受体是其中之一。GPR84通过综合表达序列标签数据库搜索的方法发现的,然后从GPR84编码的人外周血中性粒细胞中克隆和表征。GPR84主要表达于骨髓细胞,包括单核细胞、巨噬细胞、中性粒细胞、嗜酸性粒细胞、佛波酯激活的外周血单核细胞和位于中枢神经系统的小胶质细胞。链长为9-14饱和中链脂肪酸(MCFAs)是GPR84的激动剂,参与Gai信号传导,通过抑制腺苷酸环化酶来减少环磷酸腺苷(cAMP)的产生。
然而,最有效的MCFA——癸酸 1 (Fig1)以及MCFA氧化代谢产物,3-羟基十二烷酸 2 仅表现出微摩尔效力,无法招募β-arrestin,这与GPR84仍是孤儿受体的观点一致。淋巴细胞和脂肪细胞中GPR84 mRNA表达可以通过维生素D、炎症刺激物如LPS、肿瘤坏死因子α(TNFα)和慢性低度炎症显著上调。体外合成的激动剂激活GPR84会导致吞噬作用增强、免疫细胞迁移和细胞因子、趋化因子和其他炎症因子分泌增加。GPR84激动剂已报道可增强脂肪细胞质膜相关蛋白(APMAP)缺陷癌细胞的吞噬作用,抑制脂毒性诱导的巨噬细胞过度激活,引发细菌粘附增加,显示出抗动脉粥样硬化作用,并在线粒体代谢调节中发挥作用,表明GPR84的激活可能是有益于癌症、杀菌和代谢功能障碍。
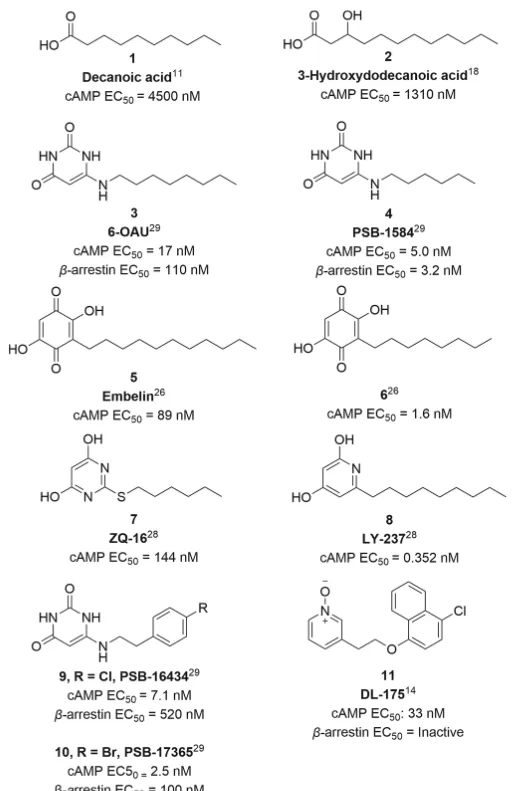
内源性的GPR84配体尚不清楚,6-辛基氨基尿嘧啶 3 ( 6-OAU )是第一个从化合物库中筛选发现的合成激动剂。该化合物由一个极性投基团和亲脂链组成的拟酸结构,是常用的阳性对照化合物,既能激活G蛋白,又能招募β-arrestin。其衍生物 4 ( PSB-1584 )在G蛋白和β-arrestin途径中显示出增强的活性。 Embelin 5 (Fig1)是一种可以激活GPR84的天然产物,发现其具有镇痛、抗肿瘤、抗炎、抗氧化和伤口愈合活性。通过对脂肪链进行修饰,化合物 6 对GPR84的激活表现出更高的效力和更好的选择性。基于160,000种化合物的高通量筛选来自中国国家化合物库,发现化合物 7 ( ZQ-16 )比化合物 6 更有效。经过SAR分析,发现化合物 8 ( LY-237 )在钙离子和cAMP实验检测中在hGPR84转染的CHO细胞中比化合物 7 和 3 更有效。
尿嘧啶衍生物 9 和 10 ( PSB-16434 和 PSB-17365 )报道对GPR84和G蛋白信号传导偏向具有增强的效力。最近有文献报道了化合物 3 和 8 与GPR84结合冷冻电镜结构,展现了GPR84激活的蛋白结构基础。在许多GPCR蛋白中,G蛋白和β-arrestin介导的信号通路已被证明具有不同的生物医学和生理作用,使得他们能够将功能效应导向特定途径。偏向激动剂可以识别具有更高功效并减少脱靶副作用的化合物,开发偏向激动剂已成为一个日益活跃的研究领域。
牛津大学使用定量结构-活性关系(QSAR)模型进行虚拟筛选,进行初步的SAR分析得到了化合物 11 ( DL-175 )。在检测cAMP积累抑制的实验测定中,显示出与化合物 3 相当的效力。然而,在测定最高浓度为60 μM下,它对β-arrestin招募没有任何影响,表明对G蛋白信号传导存在显著偏向。与化合物 3 相比,化合物 11 不能促进M1极化的U937巨噬细胞的趋化性,表明GPR84驱动的吞噬作用和趋化作用可以分开,并且诱导较少趋化性的偏向激动剂可能潜在的减少体内副作用。
此外,化合物 7 可以诱导GPR84在两个苏氨酸残基(Thr263/Thr264)上的磷酸化,而化合物 11 没有,且在GPR84受体中引入Arg172Ala突变不影响化合物 11 的活性,同时化合物 7 的活性丧失,这表明它们可能具有不同的结合模式。 然而,在小鼠肝细胞代谢实验中,化合物11会快速代谢(t1/2<10mins),导致其不能作为体内工具化合物。 由于GPR84体外和体内偏向信号传导的影响仍有待认识和理解,因此需要开发具有适合体内研究的药代动力学特征的偏向激动剂。牛津大学对化合物 11 进行分子优化成合适的体内工具化合物,用于临床前模型中GPR84疾病生理学研究。经系统的SAR分析,最终开发出具有适当选择性和ADME的GPR84有效、高度G蛋白信号偏向的激动剂 68(OX04528) 和 69(OX04529) 的体内研究。
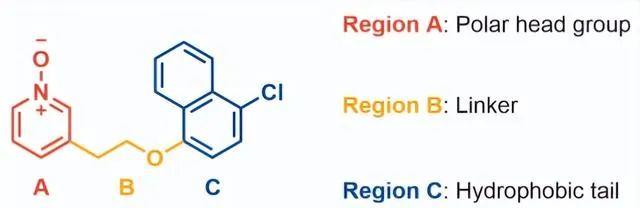
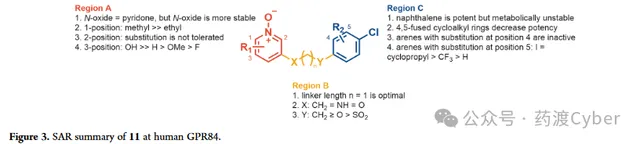
分子优化围绕GPR84偏向激动剂 11 的化学反应以形成适合体内研究的分子改造策略,牛津大学首先试图了解化合物 11 的代谢倾向。11与小鼠肝微粒体(MLM)和小鼠肝细胞一起孵育表现出快速代谢,t1/2分别为13.8mins和小于10mins。化合物 11 与小鼠肝细胞一起孵育60分钟,并使用LC-MS/MS对所得代谢物进行表征:检测到代谢物中有76%显示单氧化,其中8%形成葡萄糖醛酸结合物;12%的代谢物是二羟基化,其余的12%是未鉴定的代谢物。鉴于代谢物情况,推测氧化主要发生在萘基团,因为萘酚和二氢二醇的氧化代谢较为常见。根据SAR分析以及对化合物11代谢物的认识,该结构被分为三个区域(Fig2),即疏水尾部(区域C)、连接基团(区域B)和极性头基团(区域A)分别优化。
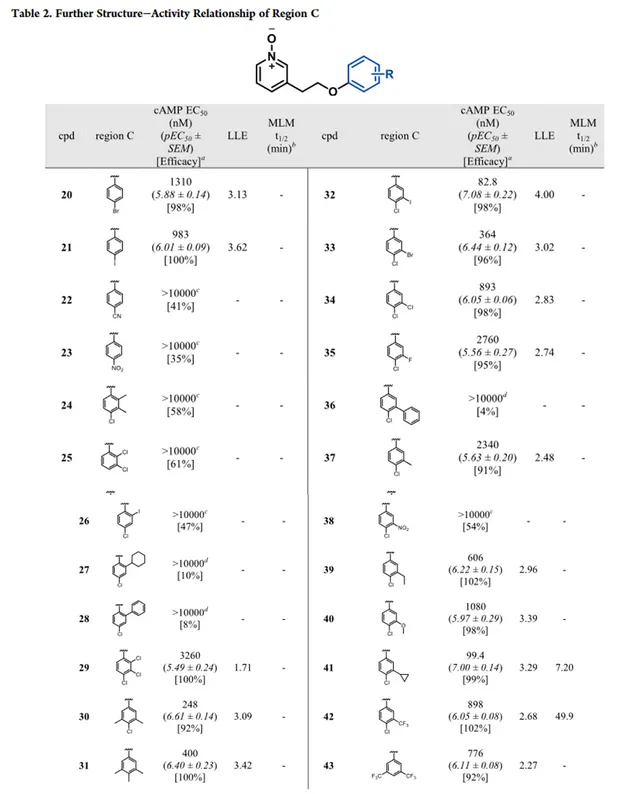
C区SAR分析
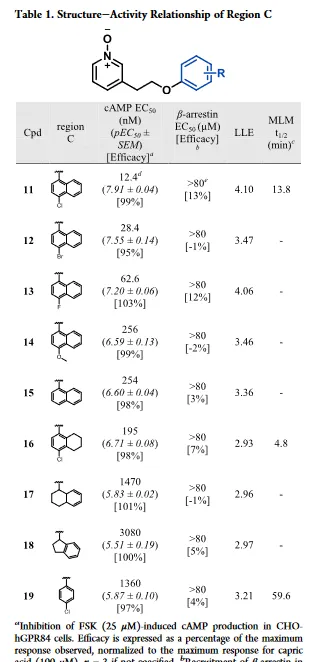
最初对化合物 11 的C区进行结构修饰,首先解决代谢不稳定的问题,如表1所示。通过抑制CHO-hGPR84细胞中毛喉素诱导的(FSK诱导)cAMP产生来测量GPR84激动剂的效力(EC50和pEC50±SEM)。通过优化分子的亲脂性来提高化合物的体外活性,还检测了亲脂配体效率(LLE)。去掉4-氯取代基或用卤素、给电子基团或氢替换4-氯取代结果说明了对卤素的偏好。化合物 16 和 17 的效力降低15至118倍, 说明平面且为芳香基团是优选片段 。与化合物 17 相比,化合物 18 的效力降低,说明需要芳香基团。
对氯苯基取代的化合物 19 与 11 相比,其效力降低110倍。尽管与 11 相比,化合物 16 和 19 效力降低,但在代谢稳定性研究实验发现,由于对氯苯基取代有利于代谢稳定性的提升( 19 ,MLM t1/2=59.6mins), 这与之前假设一致,即代谢主要位点是萘基 。由于效力降低且不存在其它亲水基团,结构改造的化合物没有高于化合物 11 的LLE。所有活性化合物与阳性对照物(癸酸)在cAMP实验中相比都是完全激动剂。全部化合物在β-arrestin实验中都没有活性(如表1),这说明 11 的衍生物在GPR84上具有一致的G蛋白信号偏倚。 根据化合物19的实验结果,进一步探索C去取代芳烃以维持代谢稳定性并增强效力。
进一步研究C区的苯基取代,设计并合成了对位取代衍生物(表2)。对位卤代衍生物 20 和 21 的效力与化合物 19 相当,与萘系列一致。对位取代的化合物 22 和 23 表现出效力显著降低,可能是由于氰基和硝基降低了芳烃电子云密度不利于氢键受体形成,或降低了疏水性。邻位取代的衍生物 24-29 效力降低或减弱。与化合物 25 和 31 相比,化合物 29 和 30 效力增加进一步强调了卤素作为对位取代基团的重要性。
间位碘取代的化合物 32 在LLE增强的情况下,效力提高20倍,说明适当的间位取代衍生物的有可能增强效力。不同的间位取代衍生物( 33-40 )效力降低进一步了解空间效应和活性之间的关系。将卤素从碘取代的 32 改为空间阻碍较少的溴 33 、氯 34 和氟 35 ,活性逐渐降低。随着受阻的3-苯基取代36的效力显著降低。而较小的3-甲基、3-甲氧基和3-乙基化合物活性表现为乙基>甲氧基>甲基。间位硝基取代的 38 没有表现出活性,而3-环丙基取代的41效力显著提升,但相比于 32 ,LLE略有下降,表明间位取代需要适当平衡空间位阻效应和分子亲脂性。
然而,在MLM研究中,发现化合物41快速代谢(MLM t1/2=7.2min),这可能是由于环丙基先被氧化所致。4-Cl-3-CF3取代的 42 和3,5-diCF3-取代的 43 与化合物 19 相比显示出效力增强,且化合物42表现出良好的代谢稳定性(MLM t1/2=49.9min)。与阳性对照癸酸相比,cAMP实验中发现所有活性化合物都是完全激动剂,并且在招募β-arrestin没有检测到活性,这与化合物 11 的结果一致。
B区SAR分析
接下来牛津大学研究连接子部分(B区),以探讨链长、杂原子的引入/去除以及所选官能团的耐受性的影响(表3)。
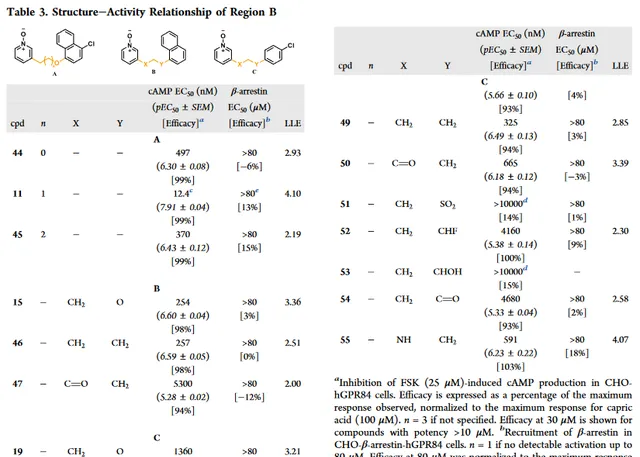
从化合物 44 、 45 和 11 相比较,很明显,连接子的长度在3个原子时是最佳的。化合物 46 和 11 比较,说明另一个连接子不是必需的且可以用烷基连接子代替。但是,换成烷基连接子可能会降低LLE。与化合物 15 和 46 相比, 47 的效力下降20倍,表明引入羰基削弱效力。与化合物 19 相比,由于不同位置的醚键连接子 48 导致效力下降2倍。有趣的是,带烷基连接子 49 和 19 相比,效力增加4倍。这与萘系列 46 不一致但显示出具有保持效力和LLE的同时替代萘的潜力。
进一步替换 49 中X位为羰基得到化合物 50 ,效力降低2倍。通过改变对氯苯基衍生物上的位置(Fig3C),如砜 51 和羟基 53 效力减弱。与化合物 19 相比,氟和羰基使效力降低10倍以上。在化合物 55 的X位置引入氨基导致类似的效力,LLE高于化合物 19 ,但效力低于 49 。与阳性对照癸酸相比,cAMP实验测定所有活性化合物都是全激动剂,并且在招募β-arrestin没有检测到活性。
N-氧化物吡啶的生物电子等排
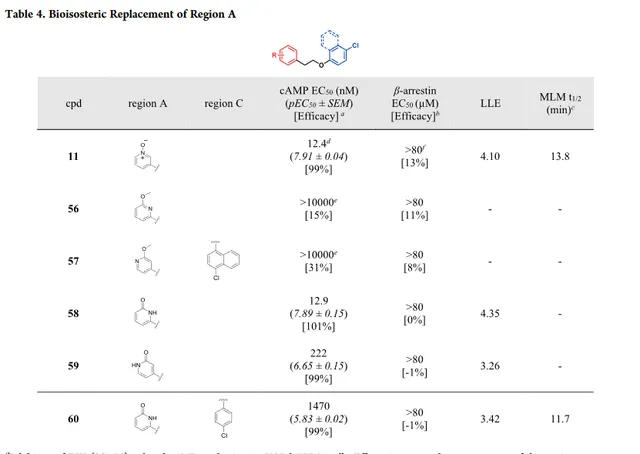
开始探索A区SAR研究之前,首先探索了非等排体替代策略,通过醛氧化酶代谢来潜在地减少吡啶N-氧化物分解。由于结构相似,氢键受体相似,吡啶酮可以是吡啶N-氧化物的生物等排体。O-甲基保护的羟基吡啶 56 和 57 在cAMP实验测定中没有活性,可能是由于甲基取代阻断氢键受体作用(表4)。取代的吡啶酮 58 在cAMP实验测定中显示完全激动活性,β-arrestin效力和LLE与化合物 11 相当,说明吡啶酮与吡啶N-氧化物具有类似的作用。4-取代吡啶酮 59 的效力降低了17倍,可能表明氢键供体N-H减弱效力。因此,将化合物 60 作为化合物 19 的生物等排体,比较代谢稳定性。 然而,与吡啶N-氧化物19相比,化合物60(MLM t1/2=11.7min)稳定性较差,说明吡啶酮部分是体内代谢稳定性差的主要来源。因此,没有进一步研究吡啶酮,开始研究吡啶N-氧化物基团修饰作为替代方案。
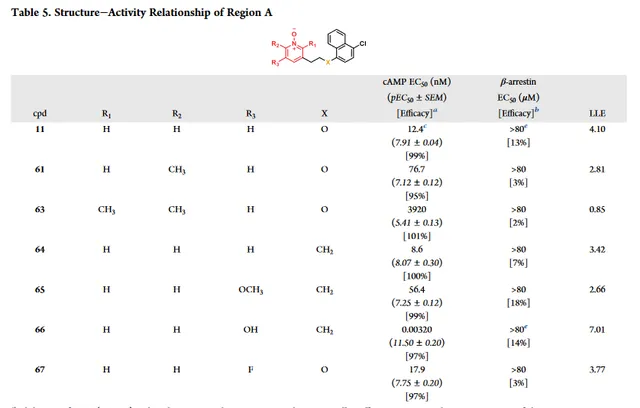
A区SAR分析
A区研究重点是通过改变R1、R2和R3的取代来增强活性(表5)。用甲基或乙基( 61 和 62 )取代R2表现出中等至大幅的效力下降,说明该位置存在空间限制。相比化合 61 ,化合物 63 在R1引入甲基活性显著降低,表明R1的取代是不可接受的。通过将醚链接子转为烷基连接子,与化合物 11 相比,化合物 64 显示出相当的效力,且LLE略有下降,这与Fig3的萘系列一致。然而,通过抑制cAMP水平,在R3引入羟基作为取代基,效力增加2000倍。与阳性对照分子癸酸相比,起到了黄曲霉素的作用,同时,在最高测试浓度(80μM)条件下,仍没有表现出β-arrestin募集作用。注意到化合物 66 的A区与 6-OAU 和 PSB-16434 内的嘧啶二酮有一定的结构相似性,使得牛津大学推测额外的羟基是否可以起到氢键受体作用,类似于 6-OAU 和 PSB-16434 的嘧啶二酮互变异构形式与GRP84的结合模式。
然而,最重要的是,在R3( 67 和 65 )导致活性降低,表明R3增加新的氢键供体实际上可能是负责增强效力。这些实验观察结果可能表明化合物 66 分别与 ZQ-16 和 LY-237 的二羟基嘧啶和二羟基吡啶互变异构体更接近。化合物 66 的这种增强效力可能是由于与GPR84中的相邻残基(与Arg172)形成新的氢键,因为化合物 11 通过独立于Arg172的机制启动GPR84激活,对于其它类似脂质配体至关重要。 随着效力增强,化合物66的LLE增加到超过5,完全再药物发现项目的可接受范围内。
SAR总结
化合物 11 周围不同的3个区域(Fig3):区域A不能耐受位置1和位置2的取代,而位置3引入氢键供体使效力提高≥1000倍,发现3个原子接头长度对于B区是最佳的;C区是主要的代谢位点,但通过用芳烃取代萘基团可以缓解。这导致活性丧失,可以通过位置5(Fig3,C区)上引入取代基并将Y更改为Cp(Fig3,B区)来缓解。
高效、有偏向且代谢稳定的激动剂设计
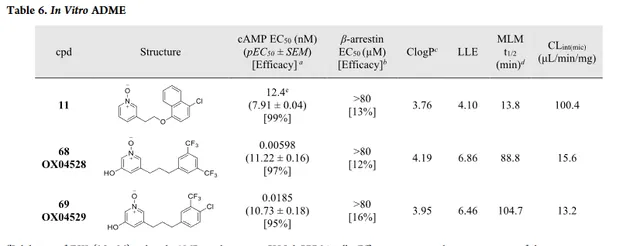
化合物 68 ( OX04528 )和 69 ( OX04529 )是通过将三个区域的主要发现合并在一起设计和合成的(表6)。
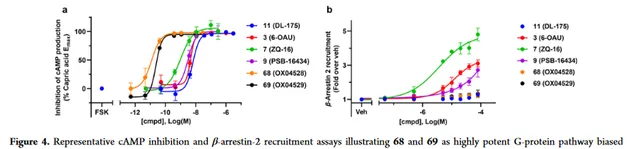
化合物 68 和 69 均表现出高效力和G蛋白信号传导偏差。一般认为,受体表达水平会影响基于细胞的测定中表观效力的测量,较高的GPR84表达水平为同一配体提供较高的表观效力值。为了避免这一潜在的混淆问题,牛津大学比较了化合物 68 和 69 在cAMP和β-arrestin招募测定的影响,以及偏倚程度不同的参考配体,比较相对效价值,其中包括 6-OAU 、 ZQ-16 和 PSB-16434 。在cAMP实验测定中,效价排序为 68>69>ZQ-16>PSB-16434>6-OAU>11 。
在β-arrestin测定中, ZQ-16>6-OAU> PSB-16434>11=68=69 。与此前报道的配体相比,化合物 68 和 69 在cAMP抑制中表现出极大的改善活性(Fig4a和表2),而两种配体在测试最高浓度下均为表现出β-arrestin募集(Fig4b)。这可能是由于与其他配体相比的不同结合模式,与之前观察到 6-OAU 和 DL175 之间药理学差异一致。这些差异需要用化合物 68 和 69 进行进一步研究。
体外ADME分析显示化合物 68 和 69 在MLM中缓慢降解(t1/2=89min和105min)。比化合物 42 和 19 代谢更稳定,表明3位羟基也可能减慢吡啶N-氧化物的代谢。随着效力增加3个对数级,化合物 68 和 69 均显示出改善的LLE>5。
新GPR84激动剂的选择性
为了确认化合物 68 和 69 诱导的cAMP产生的抑制是由GPR84介导的,对未转染的CHO-K1亲代细胞进行cAMP检测,未检测到cAMP的抑制(Fig5a)。根据先前发表的药理学证据,化合物 11 的结合被认为与 6-OAU 和 MCFA 的结合位点重叠,尽管可能具有不同的结合模式,因为受体突变研究表明GPR84 Arg172 Ala突变消除了 MCFA 和 6-OAU 的相互作用,但化合物 11 没有。化合物 11 在β-arrestin 募集激动剂和拮抗剂筛选试验中显示出 GPR84对168个人类GPCR的高选择性。 MCFA 激活GPR84表明 68 和 69 对脂质传感器FFA1和FFA4可能存在潜在的脱靶效应。感应大麻素受体2(CB2)可被癸酸1激活在微摩尔范围内,因此对这三个 GPCR 进行了选择性反筛选测试。用荧光成像酶标仪 (FLIPR) Ca2+ 测定(Fig5b-d) 68 和 69 ,显示 FFA1、FFA4和CB2无活性。
体外细胞毒性
毒性是药物开发中必须考虑的因素。因此,通过测量CHO-hGPR84细胞和CHO-K1细胞的乳酸脱氢酶(LDH)释放来检查化合物 68 和 69 的细胞毒性。在所有测试浓度(高达30μM)下孵育20小时后,化合物 68 和 69 均未显示出细胞毒性的证据。
体内药代动力学研究
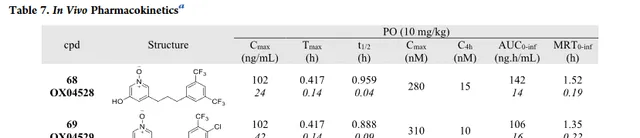
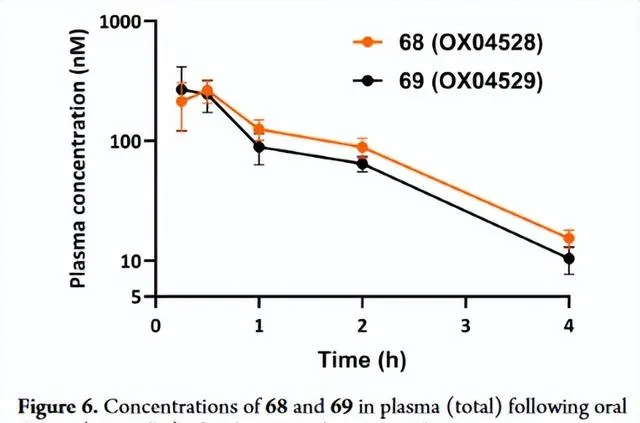
由于化合物 68 和 69 在体外测定中都表现出显著增强的效力、信号偏倚和改善的代谢稳定性,因此两者都被用于体内PK分析。两种化合物均具有口服生物利用度,并且分别具有58和53分钟的适当体内半衰期(表7)。以10mg/kg口服给药后,4小时后,两种化合物在血浆中的总浓度约为10nM,远高于细胞效力的测定值,支持它们进入体内功效研究(Fig6)。总而言之,这些数据表明这两种化合物 68 和 69 将是有用的体内探针,值得进一步研究。
结论
强效GPR84偏向激动剂 68 和 69 的发现始于用4-氯苯基替代 11 中的对氯萘(C区,Fig2)导致效力降低100倍但代谢稳定性增强。区域的进一步优化导致了活性的一些恢复并保持了MLM的稳定性。尝试用吡啶酮生物等排替代N-氧化物未能改善活性或代谢稳定性。对A区的SAR研究发现,与化合物 11 相比,添加α羟基可导致3个对数的效力增强,同时保留较高的信号偏差,这通过其在β-arrestin招募测定中的无活性来证明。为了开发具有合适的药物代谢和药代动力学特征的候选药物,设计并合成了化合物 68 和 69 。两种工具化合物都显示出极高的效力,EC50值分别为5.98和18.5pM,并且在β-arrestin测定中未检测到活性(Fig4)。
此外,这些化合物对FFA1、FFA4和CB2受体具有选择性。细胞暴露于 68 和 69 达到20小时并没有导致LDH释放,表明这些化合物在体外维持了细胞活力和低细胞毒性。化合物 68 和 69 的体外代谢研究显示MLM稳定性增强。在小鼠PK研究中,化合物 68 和 69 的口服给药显示体内半衰期分别为58和53分钟。然而,鉴于这两种化合物的高活性,血浆中的总浓度在4h时高于各自的EC50,这为进一步的体内研究提供了机会。
参考来源
J. Med. Chem. 2024, 67, 110−137
来源:药渡Cyber












