在北京市中心,3200萬元人民幣能購得一座歷史悠久、充滿傳統韻味的四合院。然而,對於某些家庭而言,這筆看似遙不可及的巨款,卻成了挽救生命的最後希望。
近期, 【NatureMedicine】 雜誌發表了一篇引人註目的研究報告,講述了 一項僅涉及一名患者的臨床試驗。而這名患者的父親,正是這篇論文的共同作者之一。

他為了拯救患有絕癥的兒子,不惜一切代價,募集了超過450萬美元(約合3200萬元人民幣)的資金,從零開始建立了一家生物技術公司,並參與了兒子的治療方案設計,逐步攻克了這一無人問津的疾病領域。
這個故事背後, 是一對父母如何將「不可能」變為「可能」的感人傳奇。
嬰兒時期遭逢不治之癥,父母誓要扭轉命運的惡作劇
麥可於2017年12月17日出生,是家中最小的孩子。然而,他的到來並沒有給家庭帶來太多歡樂,因為不久後,他的父母便發現了一些異常。
麥可的母親首先發現了問題: 他的發育明顯落後於正常水平,5個月大時仍無法擡頭或舉起雙手。他們還發現,麥可在抓握時手指似乎總是無力。

這種強烈的不安感驅使著他們開始尋求醫療幫助。經過一系列神經系統專科檢查,麥可被診斷出肌張力降低,頭顱磁共振檢查也顯示胼胝體和腦白質存在異常訊號。不久後,麥可還經歷了一次嚴重的癲癇發作。
2019年(麥可18個月大時) ,基因檢測顯示,他的AP4M1基因存在復合雜合突變,這種變異導致該基因編碼的銜接蛋白復合體4功能喪失。由此基因變異引起的疾病被稱為遺傳性痙攣性截癱50型(SPG50),這種疾病的癥狀通常在嬰兒期出現,包括全面發育遲緩、進行性小頭畸形和顱腦磁共振異常。
SPG50是一種非常罕見的疾病,且該疾病呈慢性進展,患者會因痙攣逐漸喪失運動能力,最終導致殘疾,並伴有嚴重的認知功能障礙,往往活不到成年。 目前,全球範圍內已知受該疾病影響的患者僅80人,麥可是加拿大唯一確診的SPG50患者。
麥可的父母不願放棄,他們竭盡全力尋找救治機會:與患有類似罕見疾病(如SGP47、SPG15、CMT4J等)的患者家屬交流、閱讀醫學文獻、參加學術會議……在美國的一場基因與細胞治療學會會議上,他們把兒子的疾病資訊印成「通緝令」式傳單到處分發,尋找可能了解SPG50的專家。

透過這種看似原始的辦法,他們順利地與美國一位知名的分子生物學家取得了聯系。這位專家告知麥可的父母, 利用基因編輯的方式能夠對麥可受損的基因予以修復。但糟糕的是,針對SPG50的基因治療還未被創造出來 ,而且鑒於其「一對一專屬客製」的特點,研發起碼需要300萬美元。麥可的父母毫不猶豫地變賣了所有家產。然而,僅靠家庭積蓄還遠遠不夠。他們成立了基金會,並開始聯系可以提供幫助的機構與組織——來自美國的實驗室提供了小鼠模型,波士頓兒童醫院和美國國立衛生研究院等機構提供研究支持,加拿大多倫多病童醫院的醫生為麥可設計治療方案。
很快, 善款突破450萬美元 ,相關研究也進展神速。
投資逾3200萬,展開一場僅限一人的醫學實驗
基因治療,即透過載體將包含正常基因的DNA序列匯入細胞內,使之產生目的基因的產物,從而達到治療該基因缺陷所致疾病的目的。
重組腺相關病毒載體9型(AAV9)已被證明是一種安全有效的載體,可透過鞘內給藥將基因的功能性拷貝遞送至中樞神經系統。這種給藥途徑也被廣泛套用於治療中樞神經系統疾病的臨床前和臨床研究。
目前SPG50已成為基因治療的「理想之選」。 這是因為AP4M1基因的編碼序列較短,與自身互補型腺相關病毒(scAAV)載體適配良好。透過AAV9/AP4M1構建體,能夠大量表達AP4M1蛋白,進而助力患者恢復正常的AP4M1蛋白功能。
在臨床前的研究階段,鞘內AAV9/AP4M1基因治療展現出了安全性與有效性。其中,早期幹預並且采用較高劑量能夠取得最優的治療成效 ,目標劑量被設定為10ml,包含1×10^15個載體基因組。
2021年12月30日,依據治療團隊提供的臨床前研究成果,加拿大衛生部正式批準了此項治療方案的I期臨床試驗。到了2022年2月,該臨床試驗又獲得了機構倫理委員會的批準。 就在同年3月,這場僅有一人參與的臨床試驗正式開啟。
研究人員借助腰椎穿刺鞘內註射的給藥途徑,將AAV9-AP4M1註入麥可體內,同時還為其實施了AP4M1蛋白免疫抑制治療。此次試驗的主要成果在於評估安全性和耐受性,其次要終點在於對療效的評估。
在接受治療後的12個月裏,麥可未曾出現任何嚴重的副作用,也再未發生跌倒或者癲癇發作的情況。在第3、6和12個月進行的頭顱磁共振增強、脊柱MRI平掃結果顯示,未見炎癥相關變化,腦萎縮也未出現進展。
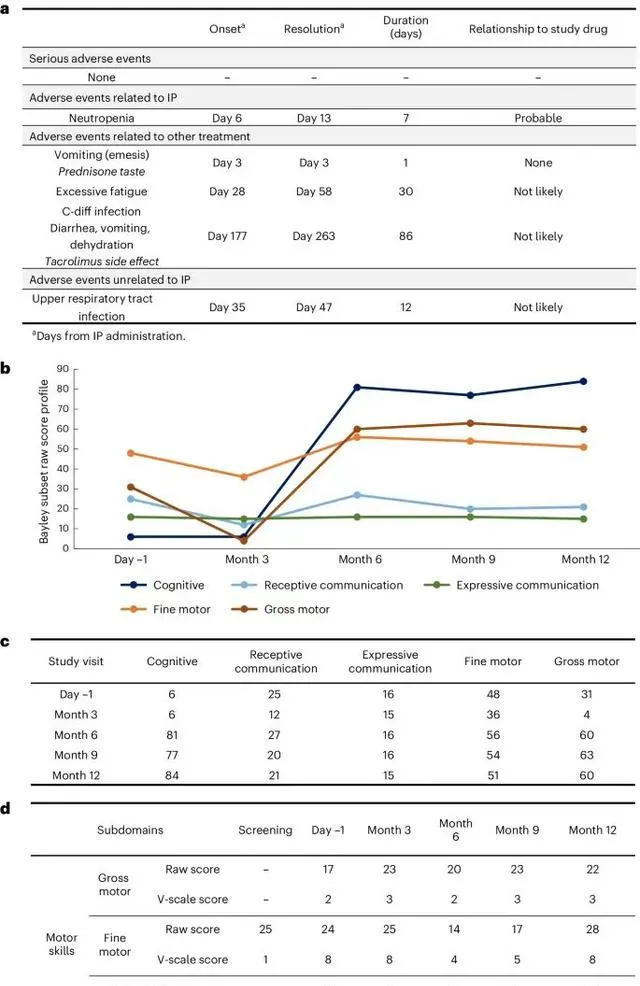
甚至在治療期的終點,麥可已經無需依賴輪椅,能夠自己用腳後跟站立。

這一令人振奮的臨床試驗結果已於6月28日在【NatureMedicine】上發表。臨床研究團隊表示,他們目前仍在持續跟蹤麥可的病情發展,不過此次試驗為基因治療在減緩或者阻止SPG50病情進展方面的安全性和有效性,提供了關鍵且重要的初步證據。
今年啟動關鍵性的第三階段臨床試驗
在兒子生病前,麥可的父親在電信和科技行業工作了25年,直至突如其來的變故改變了這一切。
「2019年4月2日,麥可被確診。一個月後,我在華盛頓與世界各地的專家會面。三天後,我們花光了畢生積蓄。一年半後,我們開始進行安全性研究。(確診的)兩年後,我們制造了這種藥物,讓麥可(確診)不到三年內就接受了基因治療。」麥可的父親說道。
從確診SPG50到讓兒子用上藥物,麥可的父母只花費了兩年半的時間,便完成了這項從「無」到「有」的突破。 這也讓他們看到,基因治療的未來潛力,可以幫助更多像麥可一樣的罕見病患者。
2023年,麥可的父親辭去工作,創辦了一家非營利性生物技術公司,專門為那些無法吸引傳統生物制藥公司的罕見病患者群體開發基因療法。
除了針對SPG50的基因治療外,該公司還研發了一款針對CMT4J(夏科-馬利-圖斯病4J型)的基因療法,這種疾病全球僅有24例 。 目前,該藥物還屬於處於臨床前階段,預計很快啟動I期臨床試驗。
2023年,生物技術行業媒體FierceBiotech將年度「突破獎項」頒發給麥可的父親——泰瑞·皮羅沃拉基斯(TerryPirovolakis)(同年同獲此獎的還有2023年的諾獎獲得者卡塔琳卡裏科) 。

FDA目前已批準針對SPG50基因療法的III期臨床試驗,該藥物被稱為Melpida(泰瑞創立的公司名為Elpida,希臘語意為「希望」。Melpida=Michael+Elpida,意味「麥可的希望」) ,將於2024年8月開始,預計納入8名患者。公司預計,接下來的臨床試驗大概需要2100萬美元 (約等於人民幣1.5億元) 。
「我已決定將畢生精力投入到拯救更多孩子上。」泰瑞說道,「這些臨床研究需要有人來買單,為此,我將放棄一切財富。」











