水平有限,公眾號轉譯的文章可能不能完全代表作者的觀點。

摘要
多倍體是真核前進演化中的重要機制,在植物界尤其普遍。然而,我們對這種現象及其對前進演化的影響的了解仍然有限。 多倍體研究的一個主要障礙是很難解開異源多倍體的起源。 由於雜交和復雜的後多倍體二倍體化過程的綜合作用造成基因組的劇烈變化和異源多倍體訊號的侵蝕, 解決異源多倍體的起源長期以來一直是一項具有挑戰性的任務。 在這裏,我們以款冬亞族(菊科:千裏光亞科)這個有趣的案例重新審視這個問題,並透過開發HomeoSorter,透過將同源基因與親本亞基因組進行定相分析,建立了一條新的網路推理管道。該管道基於先前研究的基本思路,但進行了重大更改以便解決擴充套件問題並實作一些新功能。透過模擬數據,我們證明 HomeoSorter在基因組規模的數據上能高效執行,並且在辨識多倍體模式和分配同源基因方面具有很高的準確性。 利用Phylonet的最大偽似然模型HomeoSorter和基因組規模數據,我們進一步解決了款冬亞族的復雜起源問題,這是一個物種類群(約45屬和710種),其特征是具有高堿基染色體數(主要是x=30、40)。特別是,推斷的模式得到了染色體證據的有力支持。款冬亞族被發現包含兩個具有連續異源多倍體起源的大類群:款冬亞族(主要是x=30)和類款冬亞族組(x=40)。兩個異源多倍體事件首先產生了款冬亞族,第一個事件發生在千裏光亞族的祖先(x=10)和與短舌菊屬的類群相關的譜系(很可能x=10)之間,以及由此產生的雜交譜系與山絨菊屬(x=10)的祖先雜交,並導致款冬亞族的產生。然後,在早期多樣化之後,款冬亞族的中美洲群體(主要是x=30)再次與山絨菊屬譜系一起參與第三次異源多倍體事件,並產生了類款冬亞族群體。我們的研究強調了 同源排序器和同源排序方法在多倍體系統發育學中的價值。款冬亞族具有豐富的物種多樣性和清晰的前進演化模式。和類款冬亞族群也是未來多倍體研究的優秀模型。
關鍵詞: 異源多倍體;短舌菊屬;絨安菊屬;同源排序;網狀;目標富集;款冬亞族
引言
多倍體是植物前進演化中重要且普遍的機制 (VandePeer等人2017;Clark&Donoghue2018;Fox等人2020)。它 被普遍認為是遺傳新穎性的來源(Soltis和Soltis2016)以及許多前進演化變化的驅動因素 ,包括生態席位轉移(teBeest等人,2012;Smith等人,2018)、物種多樣性的增加(Edger等人).2015;Tank等人,2015),以及災難情況下的生存(Fawcett等人,2009;Vanneste等人,2014)。目前,在植物前進演化史上已鑒定出超過180個古代多倍體事件(Ren等人,2018年;一千個植物轉錄組計劃2019年),並且估計24-35%的現存維管束物種是新多倍體(Wood等人,2019年;Mayrose等人2011;Barker等人,2016)。通常,多倍體分為同源多倍體和異源多倍體,基因組復制分別發生在同一物種內或透過不同物種之間的雜交產生(Stebbins1947;Soltis等人2014b)。雖然同源多倍體的發生率先前被低估了(Ramsey和Schemske1998;Soltis等人2007;Parisod等人2010;Barker等人2016),但 異源多倍體被廣泛認為是主要型別,並在前進演化中發揮更重要的作用 (例如,Soltis等人2003;CoyneandOrr2004;Mallet2007)。
盡管異源多倍體很普遍且很重要,但解開異源多倍體的起源長期以來一直是一項具有挑戰性的任務(Oxelman等人2017;Rothfels2021)。這首先是由於異源多倍體基因的復雜前進演化。多倍體之後通常是二倍體化過程,其中涉及復雜的基因組變化,例如基因遺失和染色體重排,隨著時間的推移逐漸侵蝕多倍體訊號(Wolfe2001;Soltis等人2015;Li等人2021)。異源多倍體結合了多個不同的基因組,通常會導致更劇烈的基因組變化,稱為「基因組休克」(McClintock1984;Bird等人2018;NietoFeliner等人2020)。此外,其他前進演化過程,包括基因重組、水平基因轉移和不完全譜系排序(ILS),可能會使異源多倍體的網狀模式進一步復混成(Steenwyk等人2023)。其中,ILS是系統發育不一致的一個特別常見的原因,並且由於它們相似的不一致模式而很難與雜交區分開來(Degnan和Rosenberg2009;Degnan2018)。因此, 異源多倍體基因往往經歷非常不同的前進演化歷史,使得很難恢復異源多倍體的真實前進演化模式。
普遍缺乏高效可靠的網路推理工具也極大地阻礙了異源多倍體的研究。傳統上,異源多倍體是根據葉綠體和核基因樹之間的細胞核不一致推斷的。測序技術的快速進步徹底改變了這一點,測序技術使我們能夠對基因組中大量未連鎖的基因座進行采樣,並恢復盡可能多的基因拷貝。這兩個方面對於辨識可靠的異源多倍體模式都至關重要(Oxelman等人,2017)。然而,大數據也對系統發育分析提出了重大挑戰,需要分析工具不僅計算效率高,而且能夠解釋復雜的前進演化過程。目前,該領域最廣泛使用的程式是Phylonet中實作的最大偽似然(MPL)模型(Yu和Nakhlep015)(Than等人,2008年;Wen等人,2018年)。它可以復雜地解釋雜交和ILS,並且通常具有很高的計算效率(Yu和Nakhlep015)。另一個近年來流行的程式是Phylonetworks(Solís-Lemus等人2017),它也是基於MPL模型,具有與Phylonet類似的效能。然而,其他程式都有很大的局限性。例如,HyDe(Blischak等人2018)可以測試特定的雜交假設,但無法估計明確的前進演化歷史。PhyDS(Edger等人2019)只需計算異源多倍體等位基因的姐妹即可推斷親本譜系。PhyLiNC(Allen-Savietta2020)和NetRax(Lutteropp等人2022)犧牲了ILS以實作更快的計算速度。這些計劃無疑過度簡化了潛在的前進演化過程,而且它們的結果通常不太令人信服。
最近,異源多倍體系統發育學的一個新興方向是對整個基因組中的等位基因進行定相,即將同源物分配給其親本亞基因組(Oxelman等人2017;Rothfels2021)。除了解決異源多倍體的親本問題外,該策略最有趣的特征是它可以產生分叉的多標記樹(而不是網路),在該樹上異源多倍體由多個亞基因組代表,這些亞基因組與各自的潛在祖先細胞聚集。透過分階段同源物和分叉樹,我們可以進一步進行一些以前對於異源多倍體來說困難或不可能的下遊分析(例如,網狀事件的年代測定),只需使用現有的成熟工具(例如,BEAST)。
提出的同源排序方法(Bertrand等人,2015年;Oberprieler等人,2017年;Lautenschlager等人,2020年;Šlenker等人,2021年;Nauheimer等人,2021年;Freyman等人,2023年;Leal等人,2023年)),Oberprieler等人的觀點。(2017)看起來簡單而有研究前景。該方法使用排列策略和物種樹重建方法將同源物分配給親本亞基因組,並最終生成整體多標記物種樹(Oberprieler等人2017)。該方法由三個步驟組成。首先,它為每個基因和多倍體生成所有可能的等位基因組合,並使用Phylonet中最小化深度合並的方法(MDC;Yu等人2013)對每個組合進行樹重建。最小化深度合並的組合被認為最適合數據並被選擇。然後,基於這些選擇,該方法為每個多倍體的所有基因生成所有可能的亞基因組組合,並執行相同的樹重建以選擇最佳組合。最後,將所有多倍體樣本的選擇組合起來,產生總體最優的多標記物種樹。Oberprieler等人使用這種方法。(2017)成功解決了具有復雜時間場景和不同型別多倍體的模擬和經驗案例。然而,由於在第二步中對所有基因的所有可能的亞基因組組合進行了詳盡的搜尋,該方法只能適用於小型數據集。對於大型數據集,計算非常昂貴甚至不可能,因為子基因組組合會隨著基因數量的增加而呈指數增長。Lautenschlager等人(2020)試圖透過開發AllCoPol來解決這個問題,采用啟發式演算法來提高計算效率。然而,問題仍然存在,因為他們仍然試圖同時計算所有基因座。預計進一步的發展會將這種有方法被套用於大型數據集。
在這項研究中,我們透過來自菊科千裏光族的款冬亞族的有趣案例來探討異源多倍體系統發育問題。千裏光科是菊科中最大的族群之一,約有150個屬和3500個物種,分布幾乎遍及世界各地(Nordenstam,2007;Nordenstam等人2009)。該部落的一個顯著特點是多倍體發生率極高,約80%的物種是多倍體(Ren等,2021)。款冬亞族作為第二大亞族,是千裏光亞科中最突出的多倍體類群。它包括約45屬和710種,主要分布在東亞和美洲(Nordenstam2007;Nordenstam等人2009)。除了3個小屬(黏子菊屬、腋絨菊屬和細葉墊菊屬)有8個物種的基本染色體數x=9(很少為7)外,所有物種均基於x=30或其衍生物(Nordenstam)1977年;諾登史坦等人,2009年。值得註意的是,五個屬(赤道菊屬、絨安菊屬、南赤道菊屬、藤蟹甲屬和擬絨安菊屬約164個南美洲特有種(Nordenstam,2007),統稱為類款冬亞族類群(Jeffrey1992),具有更高的基礎染色體數x=40(Robinson等人1997)。
在之前的一項研究中,我們開發了1104個單/低拷貝核(SLCN)基因用於千裏光亞科的系統發育學研究(Ren等人2021),並納入了一些款冬亞族物種進行測試。與二倍體千裏光亞科物種通常每個基因僅恢復一個序列相反,款冬亞族樣品通常具有兩個或三個旁系同源拷貝。此外,對於大約一半的基因,款冬亞族序列形成兩個或三個不同的、強有力支持的前進演化枝,每個樣本的旁系同源物通常均勻地分成不同的前進演化枝。在這些分支中,一個似乎總是與山絨菊屬聚集在一起,另外一兩個與千裏光族、千裏光亞族、短舌菊屬的類群和頭狀千裏光屬的美德利 - 伍德氏頭狀千裏光(Caputia medley-woodii (Hutch.) B.Nord.))有不同的關系。這些結果強烈表明款冬亞族的異源多倍體起源。然而,該研究(Ren等人2021)僅旨在選擇SLCN基因,並沒有包括足夠的款冬亞族代表。此外,與短舌菊屬的類群(另一個基於x=30的潛在異源多倍體群體)的關系非常復雜,需要更仔細的檢查。因此,我們沒有在該研究中進一步討論該問題。
這項研究有兩個主要目的。首先是為異源多倍體系統發育學 提供一種新的高效同源排序程式 。這是透過重塑Oberprieler等人的方法來完成的。(2017)解決擴充套件問題並實作一些新功能。第二個目標是自信地 解決款冬亞族的異源多倍體模式。 為了實作這一目標,我們透過合並許多新樣本來擴充套件我們之前的工作(Ren等人2021),以更好地涵蓋千裏光亞科的多樣性。使用基因組規模數據(1104個SLCN基因;Ren等人,2021)、我們的新程式和Phylonet的MPL模型,我們徹底研究了款冬亞族的起源。
材料與方法
分類單元采樣
我們首先采用了之前研究中的26個物種的序列數據(Ren等人2021)。這些包括千裏光族主要分支的24個物種,多榔菊族的狹舌多榔菊(Doronicum stenoglossum Maxim.),金盞花族的金盞花(Calendula officinalis L.)。千裏光族的串珠千裏光(Curio rowleyanus (H.Jacobsen) P.V.Heath)和向日葵族的向日葵(Helianthus annuus L.),它們是我們之前基因選擇的基礎(Ren等人,2021)。在這裏,我們另外采樣了29個物種,以更好地涵蓋千裏光亞科的多樣性。特別關註二倍體屬(即具有2n≤20任何可靠染色體報告的屬),這些屬的采樣相對密集,以便更準確地確定款冬亞族的親緣關系。這29個物種包括代表一個重要基部份支(在亞伯達菊屬(墊菊屬 Cass.)之後第二個分化)的板葉卡佩利奧菊(Capelio tabularis (Thunb.) B.Nord.),該分支在我們之前的研究中未曾涉及;有莖頭狀千裏光(Caputia scaposa (DC.) B.Nord.),這是一個孤立屬分支中的第二個物種;來自千裏光亞族15個另外屬的15個物種;來自短舌菊屬類群另外3個屬的3個物種;代表類款冬屬組的裸葉金雞菊(Gynoxys psilophylla Klatt);以及來自款冬亞族另外8個屬的8個樣本。除此之外,還從菊科的其他主要譜系中取樣了19個其他物種,以進一步測試和過濾家庭環境中選定的SCLN基因。本研究總共包含76個樣本,詳細資訊參見Dryad上的補充表S1(https://doi.org/10.5061/dryad.573n5tbf7)。
實驗方案
Ren等人選擇的1104個SLCN基因。(2021)用於本研究。Ren等人采用的26個樣本。(2021)已經有了序列。串珠千裏光和向日葵的序列分別檢索自OneKP(www.onekp.com;程式碼:BMSE)和GenBank(登入號:GCA_002127325.1;Badouin等,2017)。在這裏,我們生成了48個新樣本的數據。靶標富集的實驗室方案嚴格遵循Ren等人的實驗方案。(2021)。使用改良的CTAB方案(Doyle和Doyle1987)從矽膠幹燥的葉子或植物標本中提取基因組DNA,並在QsonicaQ800R2(美國康乃狄克州紐敦)中超聲處理至300-400bp。使用NEBNext試劑盒(Ultra II DNA 文庫制備試劑盒E7645和多重寡核苷酸E6440;新英格蘭生物實驗室,IP交換機,麻薩諸塞州,美國)制備DNA文庫,然後使用Ren等人使用的相同客製探針進行溶液內序列捕獲。(2021),其是基於單獨的外顯子設計的,參數為120聚體誘餌和6X平鋪密度。按照制造商的方案進行八個捕獲反應,每個反應從六個DNA文庫中均勻混合總量500ngDNA,雜交在65°C下保持40小時,最後進行14個PCR擴增迴圈。使用Qubit3.0熒光計(賽默飛世爾科技,Waltham,麻薩諸塞州,美國)和配備高靈敏度D1000ScreenTapes的Agilent2200TapeStation系統(安捷倫科技公司,Santa Clara,加利福尼亞州,美國)評估DNA濃度並對文庫進行定量。在Novogene(美國加利福尼亞州沙加緬度)的Illumina HiSeq 4000平台(美國加利福尼亞州聖地亞哥)上使用雙端150bp進行測序。
基因組裝
原始讀取首先在Trimmomatic 0.38版本中進行清理(Bolger等人2014),透過刪除最多兩個不匹配的接頭、30的回文剪輯閾值、10的簡單剪輯閾值、檢測甚至單個接頭堿基,並保留短片段的兩個讀數。前導5個和尾隨15個低品質堿基也被去除,並以4個堿基視窗大小和15的平均品質閾值進行滑動視窗修剪。短於40的清理讀數被進一步丟棄。生成的配對讀數在FastQC 0.11.9版本中合格(可在https://www.bioinformatics.babraham.ac.uk/projects/fastqc/獲取)並用於基因組裝。
1104個目標基因在HybPiper 1.3.1版本中組裝(Johnson等人2016)通常使用預設設定。它包括使用BWA 0.7.1版本(Li和Durbin2009)將讀數分配給目標基因,SPAdes 3.12.0版本(Bankevich等人2012)用於組裝重疊群,以及Exonerate 2.2.0版本(Slater和Birney2005)用於將重疊群與參考比對並確定外顯子-內含子邊界。
SLCN基因的旁系同源性和過濾
就像我們之前的研究(Ren等人2021)一樣,許多基因和樣本在基因組裝過程中都發出了旁系同源警告。繼Ren等人之後(2021),我們重新評估了家族背景下的所有基因,並排除了那些在不同部落中表現出同源性的基因。檢索了HybPiper辨識的所有潛在旁系同源物。對於沒有旁系同源警告的樣本,我們使用最長的從頭組裝的重疊群,而不與其他重疊群連線,從而降低了嵌合體的風險。這些序列與MAFFT 7.407版本中的串珠千裏光和向日葵的序列進行比對(Katoh和Standley2013)采用L-INS-I精度導向方法,然後在trimAl 1.4版本中進行修剪 (CapellaGutiérrez等人2009)具有自動嚴格模式。然後進一步排除剩余核苷酸少於300個的序列(不計算缺口)。各個數據集已送出至RAxML 8.2.11版本(Stamatakis2014)用於最大似然(ML)分析,全部采用GTR+GAMMA模型和20次搜尋重復。使用ETE3工具包評估基因樹(Huerta-Cepas等人,2016)。僅保留那些所有部落在單個基因樹上都是單系的基因。
此外,為了盡量減少缺失數據的潛在影響,進一步排除了缺失程度較高的基因。具體來說,我們透過保留超過41個(>75%)千裏光科樣本(包括串珠千裏光)、19個(>75%)款冬亞族和短舌菊屬的類群樣本以及11個(>50%)非千裏光科樣本來過濾基因(包括向日葵)。這裏可能值得註意的是我們只采用Ren等人的26個物種的原因。(2021)用於這項研究。Ren等人使用的48個物種中(2021),27屬於Ligularia-Cremanthodium-Parasenecio(LCP)復合體(Liu等人2016),它是款冬亞族的一個主要分支,主要分布在東亞(Pelser等人2007;Ren等人2021)。該復合體被用作範例來測試所選SLCN基因在通用和特定水平上的系統發育效用,並且采樣相對密集。如果包含所有27個樣本,則該基因過濾中的偏差將是不可避免的,與其他款冬亞族群體相比,LCP復合體顯著偏向於LCP復合體。因此,我們只使用了五個樣本來代表這個復合體,其中五個最大的屬各一個。這一數位與款冬亞族其他主要類群的抽樣數量相似(3-5個,但雌性類群只有一個可用)。最後,我們的過濾結果留下了727個基因。這些是以下所有分析的基礎,以下稱為G727數據集。
千裏光族主要譜系的鑒定
由於網路推理的計算量很大,因此我們必須進行二次采樣才能進行分析。首先生成了一個初步的系統發育框架,以驗證先前確定的千裏光亞科的主要譜系(Pelser等人2007,2010)。這裏使用了G727數據集,但丟棄了所有潛在的旁系同源(即,僅使用了HybPiper建議的「主」副本;以下稱為G727_Hyb數據集)。超矩陣和多物種合並方法都被用來推斷系統發育。對於超級矩陣方法,使用與HybPiper一起分發的fasta_merge.py指令碼連線基因,然後在RAxML中進行分析。各個基因被分配為單獨的分區,全部采用GTR+CAT模型。使用具有100個引導復制的快速引導演算法來評估節點支持。對於多物種合並方法,首先使用RAxML與GTR+GAMMA模型和100次快速引導復制生成單個基因樹。然後,跟隨Zhang等人(2018)和Ren等人(2021),Newick Utilities中支持度低於30%的分支被折疊(Junier和Zdobnov2010),以提高物種樹推斷的準確性。使用ASTRAL-III5.7.3版本中總結了物種樹(Zhang等人2018),節點支持透過局部後驗機率來測量(SayyariandMirarab2016)。
系統網路分析
首先使用Phylonet 3.8.2版本中的MPL模型(Yu和Nakhlep015)研究了款冬亞族的網狀前進演化(Than等人,2008年;Wen等人,2018年)。該模型可以推斷顯式網路,同時考慮ILS,並且可以處理相對較大的數據集(Yu和Nakhlep015)。然而,考慮到千裏光科中多倍體的普遍存在,本研究中約70%的千裏光科樣品是多倍體,並且可能具有獨立的起源。在一次分析中不可能分析所有這些。因此,我們采取了一步一步的方法,每一步都進行不同的子采樣,以最終闡明款冬亞族的起源。
這裏使用了G727數據集的各個基因樹,全部使用Newick Utilities進行植根。此外,支持度低於30%的分支被折疊,這一策略被證明可以改善ASTRAL分析中的樹推理(Zhang等人2018;Ren等人2021),並且在這裏也進行了嘗試。
第一個分析(以下稱為Phylonet_A1)是為了了解千裏光亞科的基本網狀圖案。選擇了22個樣本來代表上述千裏光科的主要譜系,包括代表四個不同二倍體譜系的四個物種(亞伯達菊屬的凹緣亞伯達菊(墊菊屬 emarginata (Gaudich.) Cass.)、卡佩利奧菊屬的板葉卡佩利奧菊(Capelio tabularis)、切爾索多馬屬的白色切爾索多馬(Chersodoma candida Phil.)和頭狀千裏光屬的美德利-伍德氏頭狀千裏光(Caputia medley-woodii (Hutch.) B.Nord.))、短舌菊屬聯盟的莫羅短舌菊(短舌菊屬 monroi (Hook.f.) B.Nord.)、代表款冬亞族六個主要組的六個物種,以及千裏光亞族的11個物種(從二倍體到十倍體)。ETE3用於修剪單個基因樹。由於很難確定真實的網狀結構數量(Cao等人2019),因此進行了六次測試,分別允許1到6個網狀結構事件。任何測試均未指定雜交物種(即未使用「-h」標誌)。
由於Phylonet_A1的結果表明款冬亞族譜系出現在現存千裏光亞科的多樣化之前,因此千裏光亞科多倍體可能與款冬亞族的起源沒有直接關系。為了進一步檢驗這一假設,進行了14項額外分析(Phylonet_A2–A15),每項分析都研究了千裏光亞科的多倍體樣本。類群采樣在Phylonet_A1的基礎上進行了調整,非千裏光亞科樣本不變,千裏光亞科采樣替換為其全部二倍體樣本加1個多倍體樣本。對於每次分析,允許網狀結點的數量最多為所研究物種的倍性水平的一半(即比潛在網狀化事件多一個)。例如,對於番千裏木屬,2n=10x=100,通常預計會出現最多四個網狀事件,我們進行了五次測試,允許最多五個網狀結構以獲得更可靠的結果。此外,與Phylonet_A1不同的是,所研究的千裏光亞科多倍體需要透過「-h」標誌指定為雜種;否則,程式將始終首先推斷款冬亞族的網狀結構。
在確認千裏光亞科多倍體與款冬亞族的起源無關後,我們排除了所有千裏光亞科多倍體,並更徹底地研究了款冬亞族的網狀模式(Phylonet_A16)。對於分類單元采樣,我們保留了四個二倍體物種來代表千裏光亞科,並將款冬亞族和短舌菊屬的采樣擴充套件到每個主要組的三個樣本,但雌性類組中只有一個可用。進行了六次測試,分別允許一到六次網狀事件,並且所有測試都沒有指定雜交物種。
在所有上述分析中,六倍體短舌菊屬的類群尚未推斷出任何雜交訊號,並且款冬亞族五個主要類群的親本譜系通常與該類群直接相關。為了證實這種模式,我們針對短舌菊屬的類群(Phylonet_A17)進行了額外的分析。分類單元采樣是根據Phylonet_A16進行調整的。短舌菊屬的類群的四個樣本都包括在內。綿毛千裏光,千裏光屬的第二個物種,與短舌菊屬的類群關系最密切。款冬亞族的樣本被簡化為五個物種,除了不相關的雌性類群外,每個主要類群各有一個。類似地,進行了六次測試,分別允許一到六次網狀事件,並且所有測試均以短舌屬聯盟和節肢亞科設定為雜交種。
為了獲得可靠的結果,所有上述測試重復十次,每次搜尋重復20次(「-x20」)。具有最高對數偽似然值的結果被認為最適合數據。
同源物排序
為了促進同源分選方法在基因組時代的使用,並為款冬亞族的網狀前進演化提供額外的證據,我們基於Oberprieler等人的基本思想開發了一種新的流程(2017)但有重大變化。如上所述,Oberprieler等人(2017)試圖一次計算所有基因的所有子基因組組合,這對於大型數據集來說是不切實際的,因為子基因組組合會隨著基因的增加而呈指數增加。在這裏,我們引入了一種分層方法,其中我們逐漸將基因分組在一起,並在每一步中為每個組選擇局部最佳組合,直到所有基因最終組合以確定總體最佳亞基因組組合。
除了這個改變之外,我們還做了一些其他的重大調整。首先,我們 使用ASTRAL-III 5.7.3版本(Zhang等人,2018)和ASTRAL-Pro 1.1.2版本(Zhang等人2020)代替用於樹推理的MDC模型 。與簡約的MDC模型相比,ASTRAL程式在解釋ILS方面更加復雜(Zhang等人2018,2020)。盡管Phylonet也有復雜的模型(例如最大似然和MPL),但ASTRAL程式仍然是首選,因為它們的計算速度更快。其次,我們放棄了Oberprieler等人(2017)方法的第一步,即單獨分析基因,並且對於每個基因和樣本,每個亞基因組最多只允許有兩個等位基因。相反,我們 將基因樹分成小組,並將這些小組視為最基本的分析單位。 此外,將等位基因分配給亞基因組沒有限制(即,一個或所有等位基因可以分配給同一亞基因組);這可能有助於減輕前進演化噪音或PCR擴增和/或測序過程中引入的一些錯誤的潛在影響。第三,在分析多個多倍體時,Oberprieler等人。(2017)分別研究了它們,僅在最後一步合並結果。然而,我們試圖盡 可能全面地分析它們(見下文),因為它們的前進演化歷史可能密切相關,應該作為一個整體來考慮。
下面我們更詳細地描述了管道。首先,對輸入的個體基因樹進行格式化,主要是為多倍體種質附加獨特的基因索引,防止在生成等位基因/亞基因組組合過程中發生混合。然後,透過排除所有其他多倍體,對每個研究的多倍體的輸入樹進行修剪。第三步包含我們管道的核心過程,圖1中透過一個簡單的範例進行了說明。它由多個子步驟組成,每個子步驟都涉及兩輪ASTRAL分析。在第一個子步驟中,根據限制每組中每個多倍體的等位基因總數的標準(即「--allele」標誌),將各個基因樹分成小組。例如,圖1中的前三棵樹都具有一個多倍體(紅色)的兩個等位基因和第二個多倍體(藍色)的兩個等位基因。如果「--allele」設定為「4」,則只有前兩棵樹可以分組在一起,因為如果也包括第三棵樹,第一個多倍體的等位基因總數將超過限制。對基因進行分組後,我們為每組中的每個多倍體生成所有可能的等位基因組合,並使用ASTRAL-III為它們重建樹。選擇最終歸一化四重奏得分最高的前幾個(「--最佳」)組合(如果多個組合得分相等,則隨機選擇一個或多個組合)。然後在第二輪ASTRAL分析中對它們進行重新評估,並考慮所有多倍體。所有可能的亞基因組組合都是透過第一次ASTRAL分析中選擇的所有多倍體組合的排列生成的,然後使用ASTRAL-III進行計算。選擇對整體最優做出貢獻的一個。
在第二個子步驟中,每幾個基因組(「--every」)被匯集在一起作為一個更大的組。對於每個較大組中的每個多倍體,所有可能的亞基因組組合都是根據前一個子步驟的結果生成的,並在ASTRAL-Pro中分析前幾個(「--最佳」)組合。然後在ASTRAL-Pro中重新評估選定的組合,以確定有助於整體最佳化的組合。圖1所示的範例很簡單,整個過程只需要兩個子步驟。對於大型數據集,第二個子步驟可能需要重復多次才能最終將所有基因分組在一起以生成最終的最佳多標記物種樹。
應該註意的是,我們在第一個子步驟中使用了ASTRAL-III,在其余子步驟中使用了ASTRAL-Pro。這是因為這兩個程式雖然基於相似的演算法,但執行起來卻截然不同。根據我們的測試,ASTRAL-III在辨別競爭性多標記樹之間的差異方面更加敏感,這在最初的等位基因到亞基因組分配中尤為重要,但其計算效率隨著等位基因的增加而大大降低。ASTRAL-Pro可以更有效地處理大型數據集,因此被用作替代品。
由於我們的新管道的一些缺點和隨機問題(參見討論),不同執行的結果可能會有所不同。可以執行多次重復(「-r」)來總結主要模式並評估穩健性。此外,為了獲得更有說服力的結果,我們開發了兩種調整樹輸入的策略,一種是透過打亂輸入基因樹的順序(「--shuffle」),另一種是透過放回隨機抽樣樹(「--引導」)。然而,排序的亞基因組在不同的重復中被隨機命名。在總結結果之前,我們需要手動辨識亞基因組並相應地重新命名它們。該管道采用Python3語言編寫,命名為「HomeoSorter」。該指令碼和更多詳細資訊可在https://github.com/ChenRen-SCBG/HomeoSorter上獲取。
HomeoSorter的模擬測試
我們首先使用模擬數據來測試新管道的效能。使用HybridSimv.1.0.1(Woodhams等人2016)總共模擬了238個數據集,這是一個可以模擬各種型別網狀結構的優秀程式。更重要的是,對於每個模擬網路,HybridSim提供了一組在隨機合並過程下生成的隨機選擇的基因樹(即HybridSim定義的「合並樹」),作為HomeoSorter測試的理想輸入。前75個數據集用於測試經常影響網路推理準確性的兩個主要因素,即父本母本貢獻(即HybridSim定義的「混合分布」)和ILS。具體來說,我們模擬了具有五種不同比例的親本貢獻(50:50、40:60、30:70、20:80和10:90;從相等到不相等)和15個合並時間(CT;或「合並」)的四倍體。每個比例的比率」,由HybridSim使用,範圍從0.1到200(從極高到低雜訊;比率與HybridSim定義的物種形成率相關,預設為1)。每個數據集由8個分類單元和200個合並樹組成。對於其他參數,我們遵循預設設定,除了關閉基因滲入,這可能會改變合並樹中親本貢獻的預設比例。特別是,我們對所有模擬使用相同的種子(「-s111」)。透過這樣做,HybridSim可以生成相同的網路,更重要的是, 可以減少模擬的隨機性,並確保測試因素之間的差異最大程度地反映在合並樹中。
接下來的15個數據集用於測試HomeoSorter在同源多倍體方面的效能。由於HybridSim無法模擬同源多倍體的系統發育,我們生成了一個簡單的分叉樹,並將兩個姐妹類群指定為同源多倍體的兩個亞基因組。同樣,每個數據集都是由8個分類單元和200個合並樹生成的。模擬了從0.1到200的CT,所有CT都具有相同的種子「111」。
其余數據集用於評估HomeoSorter在各種場景下的計算時間。測試了六個主要因素,包括所研究的多倍體數量、倍性水平、基因數量以及HomeoSorter的三個主要參數(「--allele」、「--best」和「--every」)。數據集使用20個分類單元和200至4000個合並樹進行模擬,所有樹均具有相同的「111」種子。
對於HomeoSorter分析,使用HybridSim生成的合並樹作為輸入。除非另有說明,否則均遵循HomeoSorter的預設設定。對於前75個數據集,我們進行了100次重復的改組分析,並總結了正確辨識親子關系的平均支持度。對於每個重復,我們進一步計算了正確等位基因分配的百分比。至於同源多倍體的測試,我們還對每個數據集進行了100次重復的改組分析,並總結了對同源多倍體模式正確辨識的支持(即,四倍體的亞基因組形成一個分支,然後與其最接近的親屬成為姐妹)。然而,未評估等位基因分配,因為將等位基因分配給任一亞基因組在技術上是正確的。為了評估計算時間,我們對每個數據集進行十次重復的改組分析並計算平均時間。所有分析均在配備16核Intel Xeon E5-4657L v2 CPU(2.40GHz)和256G RAM的機器上執行。為了評估計算時間,僅使用一個執行緒以獲得更準確的評估。
為了進行比較,我們還使用AllCoPol和Phylonet的兩個模型(MPL和MPAllopp)分析了前90個數據集。上面已經提到了AllCoPol和MPL。MPAllopp(Yan等人,2022年)是一種擴充套件的MDC模型(Yu等人,2013年),並聲稱能夠比Oberprieler等人(2017年)的方法更可能且更優越地處理多倍體,而無需進行排列(Yan等人,2022)。因此我們也在這裏進行了測試。Allcopol測試使用預設設定執行。MPAllopp和MPL測試均在指定和不指定多倍體的情況下執行,每個測試都有20個搜尋重復(「-x20」)。由於MPAllopp和MPL是為網路分析而開發的,對於同源多倍體,它只能推斷出一種模式,其中導致同源多倍體的兩個分支都與單個分類單元最密切相關。我們認為這是MPAllopp和MPL分析中同源多倍體的正確辨識。
款冬亞族的HomeoSorter分析
對於款冬亞族的HomeoSorter分析,我們使用了Phylonet_A16分析的相同數據集,但添加了向日葵作為外群。然而,該數據集包含19個多倍體樣本(款冬亞族和短舌菊屬的類群),如果一起分析,將在第二輪ASTRAL分析中產生大量的亞基因組組合。由於短舌屬聯盟和款冬亞族亞科的主要類群都得到形態學、細胞學和分子系統發育學證據的有力支持(Nordenstam等,2009),因此我們在這裏透過分析主要類群而不是物種來規避這個問題。這是透過將多倍體序列視為相應組的「等位基因」來完成的。例如,橐吾屬(Ligularia Cass.)、垂頭菊屬(Cremanthodium Benth.)和蟹甲草屬(Parasenecio W.W.Sm. & J.Small)是 LCP 復合體的成員;這三個屬的所有序列在此被視為復合體的「等位基因」。還應該指出的是,兩棲太平洋類群(包括黏子菊屬、腋絨菊屬和細葉墊菊屬)在款冬亞族中的特征是其基礎染色體數為x=9(很少為7),但分子系統發育證據表明,它在異源多倍體後發生分化款冬亞族(Ren等人2021)。他們也被視為六倍體譜系。該調查進行了50次改組和50次自舉重復,設定相同「--等位基因8--最佳3--每3」。手動辨識和重新命名子基因組後,我們使用DendroPy 4.5.2版本(Sukumaran和Holder2010)的sumtrees.py指令碼總結了大多數共識樹和支持值(分別用於改組和引導重復)
基於同源排序的系統發育重建
為了進一步探討HomeoSorter的實用性和缺點,我們測試了基於階段性等位基因的系統發育重建。這裏使用了50次改組重復的結果。對於每個重復,都有一個等位基因分配列表。根據每個列表,我們基於原始G727數據集構建了一個新的序列矩陣,並將款冬亞族序列重新命名為其分配的亞基因組。在某些情況下,多個序列被分配給同一個亞基因組;我們生成了嚴格的一致序列來代表亞基因組。使用超級矩陣和多物種合並方法重建系統發育,設定與上面使用的相同。對於超級矩陣方法,為每個重復生成一個串聯的ML樹,並使用sumtrees.py總結多數共識樹。對於合並方法,我們為每個重復的單個基因生成了ML樹,然後將它們匯集在一起(總共36350個樹,其中50個重復每個727個樹)以總結ASTRAL-III中的物種樹。
由於ASTRAL方法產生了與HomeoSorter分析類似的結果,但超級矩陣方法卻沒有,我們進一步測試對數據集進行過濾,以減少HomeoSorter的一些缺點的潛在影響。由於款冬亞族的單系性(包括類款冬亞族群,但不包括山絨菊屬的姊妹組D亞基因組)得到了強烈支持,因此排除了那些在ML樹上形成分支但分配給不同亞基因組的款冬亞族序列。然而,這種做法導致了額外的數據缺失。為了減輕它們對系統發育推斷的潛在影響,我們進一步排除了款冬亞族和短舌菊屬的類群樣本少於15個(<75%)的基因。然後,我們使用與上述相同的設定重新執行超級矩陣和多物種聚結分析。
結果
基因組裝和旁系同源
如補充表S1和圖S1(Dryad)所示,除了兩個DNA嚴重降解的樣本(Capelio tabularis和Hertia cheirifolia Kuntze)外,基因恢復情況非常好,平均恢復了1040個(約94.2%)基因。對於非千裏光科樣品,有千裏光屬和1008(約91.3%)。總共854起(約77.4%)收到了Paralog警告。然而,對於具有或極有可能具有2n≤20的千裏光樣本以及除一個非千裏光樣本外的所有樣本,它們有不到99個(約9%)基因接受旁系同源警告,平均有33個(約3個)%)每個樣本的基因。來自異源多倍體屬的狹錐花佩菊(Liu等人2013)是唯一收到大量旁系同源警告(199個基因)的非千裏光科樣本。相比之下,對於2n>20的千裏光科樣本,平均有223個(約20.2%)基因發出旁系同源警告,其範圍從佛羅裏達美幌菊(Arnoglossum floridanum (A. Gray) H. Rob.)的53個(約4.8%)不等。(2n=52;例如Jones1970) 到肯亞千裏光(Dendrosenecio cheranganiensis(Cotton & Blakelock) E. B. Knox)的531個(約48.1%)(n=50;Knox和Kowal1993)中的531(約48.1%)。
對單個基因的系統發育分析表明,68個基因樹中的一些部落是非單系的,這表明在部落分化之前存在潛在的基因重復。此外,309個基因存在大量缺失數據。這些基因被排除在外,以減少它們對系統發育推斷的潛在影響。然後總共保留727個基因用於以下分析。
千裏光族主要譜系的鑒定
基於G727_Hyb數據集的串聯ML(補充圖S2;Dryad)和ASTRAL(補充圖S3;Dryad)分析表明菊科的部落關系幾乎相同,這也與Mandel等人的結果相似。(2019)基於一組完全不同的核基因。然而,就千裏光族內部的關系而言,它們之間表現出一些不一致,並且進一步不同於Pelser等人生成的部落框架。(2010)使用葉綠體基因。盡管如此,在部落內可以很容易地辨識出八個主要分支/譜系,即(1)墊菊屬,(2)Capelio菊科,(3)千裏光亞科/Othonninae亞族(因為Othonninae總是巢狀在千裏光亞科內,該分支在下文中被稱為為了方便起見,稱為千裏光亞科(Senecioninae)),(4)短舌菊屬(包括線絨菊屬,短舌菊屬J.R.Forst.&G.Forst,粉蟹甲屬和黏菊木屬),(5)山絨菊屬,(6)Caputia菊科,(7)絨安菊屬,和(8)款冬亞族,不包括類款冬亞族組(以下稱為款冬亞族)。
在款冬亞族中,Pelser等人先前確定了五個主要類群。(2010)並且與它們的分布模式高度一致在這裏得到了證實。其中包括(1)東亞類群(即LCP復合體),主要為東亞特有種,以垂頭菊屬、橐吾屬、羽葉菊屬、蟹甲草屬和狗舌草屬為代表;(2)中美洲群,以番蟹甲屬、肉脂菊屬、印第安菊屬、傘蟹甲屬和頂葉千裏光屬為代表;(3)北美西部類群,以類蟹甲屬、帚蟹甲屬、覆旋花屬和四蟹甲屬為代表,(4)溫帶歐亞類群,包括山雛菊屬、蜂鬥菜屬和款冬屬,(5)兩棲太平洋類群,由黏子菊屬、腋絨菊屬和細葉墊菊屬組成。根據這些結果,我們對Phylonet和HomeoSorter分析進行了二次采樣。
系統網路分析
Phylonet_A1分析表明,當僅允許一種網狀結構時,山絨菊屬被推斷為雜種(圖2a)。然而,當允許有兩個網狀結構時,款冬亞族和絨安菊屬被認為是雜交種,其親本為款冬亞族。與山絨菊屬和千裏光亞科的祖先有關,絨安菊屬與傘蟹甲屬(中美洲款冬亞族的成員)和山絨菊屬相關(圖2b)。經過三項事件的測試,疆千裏光屬還被推斷為雜種,起源於千裏光亞科(圖2c)。當四個網狀結構被允許時,在短舌菊屬譜系和千裏光亞科的祖先之間推斷出一個新的網狀結構,並且由此產生的雜交譜系與山絨菊屬譜系雜交並產生了款冬亞族。(圖2d)。當允許的網紋增加到五個和六個時,與款冬亞族相關的網紋圖案。和絨安菊屬保持不變,但額外的多倍體千裏光亞科樣本甚至千裏光亞科本身被推斷具有雜交起源(圖2e,f)。六次檢驗的結果對數偽似然如圖2g中的柱形圖所示。這些值首先從單事件測試(-1038441)快速增加到四事件測試(-1035349),然後緩慢增加到五項事件(-1036272)和六項事件(-1035247)測試,這表明四項事件的結果最適合數據。
Phylonet_A2–A15重點關註多倍體千裏光亞科樣本,結果表明網狀結構很大程度上被推斷發生在千裏光亞科譜系之間(補充圖S4–S17;Dryad)。在少數情況下,涉及款冬亞族樣本(例如補充圖S4c、S9d和S11b;Dryad),但最終產生多倍體的遺傳機率均小於0.05,表明這些可能只是前進演化噪音。因此,千裏光亞科多倍體很可能都起源於該亞族內部,與款冬亞族的起源沒有直接關系。
Phylonet_A16主要針對款冬亞族(圖3)。在單項事件測試中,山絨菊屬仍被推斷為雜種(圖3a)。然而,隨著更多的網狀事件被允許,款冬亞族的網狀圖案逐漸消失。絨安菊屬逐漸出現,並且比Phylonet_A1中推斷的要清晰得多。在雙項事件測試中,款冬亞族推測山絨菊屬譜系和千裏光亞科的祖先之間存在網狀關系,而絨安菊屬在山絨菊屬和中美洲款冬亞族群的祖先之間也有另一個網狀事件。(圖3b)。在三項事件測試中,在千裏光亞科的祖先和短舌屬聯盟的祖先之間推斷出額外的網狀結構,由此產生的雜種譜系與山絨菊屬譜系雜交並產生了款冬亞族。(圖3c)。款冬亞族的網狀圖案。盡管在款冬亞族中推斷出額外的網狀事件,但絨安菊屬在四到六項事件測試中保持不變。(圖3d-f)。至於對數偽似然值(圖3g),從三項事件測試開始,它們變得總體穩定(三到六項事件測試分別為-2445021、-2444783、-2444446和-2444614)。
Phylonet_A17重點關註短舌菊屬的類群(圖4)。由於款冬亞族和短舌菊屬的類群都被指定為雜交種,對於單項事件測試,因此推斷它們具有共同的雜交起源(圖4a)。當允許存在兩個網狀結構時,這兩個群體被認為具有不同的起源,但具有與千裏光亞科相關的共同親本譜系(圖4b)。對於三到六項事件測試,得到的模式通常是相似的(圖4c-f)。款冬亞族推斷有兩個網狀事件和三個親本譜系。短聲聯盟仍然擁有一個親代血統,但這一血統現在與Caputia關系密切。另外兩個譜系分別與山絨菊屬和千裏光亞科的祖先有關。短舌菊屬的類群被推斷為三項事件測試(圖4c)、四項事件測試(圖4d)和六項事件測試(圖4f)有一個網狀結構,五項事件測試有兩個網狀結構(圖4d)),但五項事件測試中的一個網狀結構可以忽略不計,其邊緣具有非常低的遺傳機率(0.001;圖4d)。這四個測試推斷的親本譜系都與Caputia菊科關系最密切(圖4c-f),並且它們的對數偽似然幾乎相同,在-635640到-635636之間略有波動(圖4g)。
HomeoSorter的模擬測試
HomeoSorter對親本貢獻和ILS的模擬測試表明,對於親本貢獻相等(50:50)的情況(圖5a),當CT=0.1(表明ILS顯著)時,HomeoSorter只能在43.5時辨識出親本。%的案例。然而,隨著CT增加到0.5,機率迅速增加到87.5%,並且對於CT≥1,HomeoSorter進行了所有完美的辨識。等位基因分配顯示出類似但更為漸進的趨勢。隨著CT從0.1增加到20,正確等位基因分配的百分比從約50%增加到近100%,此後保持在100%。
同樣,在40:60(圖5b)、30:70(圖5c)和20:80(圖5d)父本母本貢獻的測試中,隨著ILS問題變得不那麽嚴重,HomeoSorter的效能迅速提高。然而,隨著親本貢獻變得越來越不平等,親本鑒定和等位基因分配的準確性或多或少地降低了。例如,在20:80親本貢獻測試中(圖5d),即使CT≥20,HomeoSorter也無法完美辨識親本,並且等位基因分配的準確性在重復範圍更廣、更多的情況下顯示出更大的差異。異常值(圖5d)。
父本母本貢獻10:90的案例有一個獨特的模式(圖5e)。正確辨識父本母本的機率首先增加到約80%(CT=1),然後下降到50%(CT=8),此後保持在50%。同樣,正確等位基因分配的平均百分比首先上升到80%(CT=6),然後下降到約50%(8≤CT≤200)。我們檢查了生成的物種樹,發現對於CT在8到200之間的數據集,四倍體的兩個亞基因組一致成為親本的連續姐妹,貢獻主要(90%),這表明HomeoSorter接近錯誤辨識異源多倍體稱為同源多倍體。
對於同源多倍體測試,對於CT≥6的數據集,所有重復均完美恢復了同源多倍體模式(圖5f)。當CT減少到4時,機率降低到96%,當CT≤2時,HomeoSorter辨識失敗(圖5f)。
使用AllCoPol、MPL和MPAllopp對上述數據集的測試結果分別顯示在補充圖S18-S20(Dryad)中。在親本比例為50:50至30:70的異源多倍體案例中,AllCoPol和MPL都表現出與HomeoSorter相似的效能,當CT≥1或2時完美辨識親子關系(補充圖S18ac、S19a-f;Dryad)。然而,與HomeoSorter相比,AllCoPol在等位基因分配方面的準確性要低得多,即使對於CT≥8的病例,也只有約77%(對比接近100%)(補充圖S18a-c;Dryad)。隨著親本貢獻變得更加不平等,AllCoPol表現出功率顯著下降(補充圖S18d,e;Dryad),而MPL通常表現良好,特別是在指定多倍體時(補充圖S19g-j;Dryad)。透過預先指定,即使在CR≥1時親本貢獻為10:90的情況下,親本也能被完美辨識(補充圖S19j;Dryad)。關於同源多倍體測試,AllCoPol在ILS較嚴重的情況下完美恢復了同源多倍體模式(CTs≥0.5vs.CTs≥4;補充圖S18f;Dryad),表現優於HomeoSorter,而MPL的效能相對較低。當未指定多倍體時,MPL在所有情況下都會失敗(補充圖S19k;Dryad)。即使有預先指定,MPL成功的案例(CR>6;補充圖S19l;Dryad)也比HomeoSorter成功的案例(CR>2;圖5f)要少。至於MPAllopp,在指定多倍體時通常效果很好,當CT≥1時正確辨識異源多倍體(補充圖S20b、d、f、h、j;Dryad),當CT≥2時正確辨識同源多倍體(補充圖S20l;Dryad))。然而,當沒有指定多倍體時,它通常效果不佳。盡管圖表上表明MPAllopp在CT≥40時成功辨識出異源多倍體(補充圖S20a、c、e、d、i;Dryad),在CT≥6時成功辨識同源多倍體(補充圖S20k;Dryad),但它應該需要註意的是,這些正確的模式只是MPAllopp同時推斷出的許多(最多103個)同樣簡約的網路/樹之一。
至於HomeoSorter的計算效率,在測試的六個因素中,時間隨著五個因素(即多倍體數、倍性水平、「--等位基因」、「--最佳」和「--every」;圖6a-e),但始終與基因數呈線性相依(圖6f)。
款冬亞族的HomeoSorter分析
正如HomeoSorter改組分析的多數共識樹所示(圖7),核心千裏光亞科的基本二倍體框架由三個譜系組成:山絨菊屬、Caputia菊科和千裏光亞科,其中後兩個譜系關系更為密切。短舌菊屬的類群的三個亞基因組分別附著在框架上。亞基因組A和B是Caputia的連續姐妹,都得到了全力支持。亞基因組C是大千裏光亞科/Caputia菊科前進演化枝的姐妹,但支持相對較弱(56%/-用於改組/引導支持)。至於款冬亞族,絨安菊屬有四個亞基因組和款冬亞族的五個主要類群。每個都有三個亞基因組。其中,絨安菊屬的亞基因組D是獨立的,與山絨菊屬是姐妹,得到充分支持,而其余18個亞基因組共同形成了三個大的、強支持的前進演化枝(亞基因組A-C;均為100%/100%),每個前進演化枝均勻地由絨安菊屬的一個亞基因組和款冬亞族五個主要類群的每個亞基因組組成。這三個分支分別與千裏光亞科 (68%/70%)、短舌屬聯盟的Caputia菊科/亞基因組A和B(38%/32%)以及山絨菊屬/絨安菊屬亞基因組D(100%/100%)聚類。特別是,在所有這三個分支中,絨安菊屬始終是中美洲款冬亞族群體的姐妹。(分別為92%/86%、100%/98%和82%/84%)。
基於同源排序的系統發育關系
我們根據排序的同源物進一步重建了系統發育。ASTRAL樹(補充圖S22;Dryad)與HomeoSorter改組分析的共識樹大致相似,但絨安菊屬的亞基因組C是款冬亞族的姐妹,而不是巢狀在其中。超級矩陣方法的共有樹(補充圖S23;Dryad)顯示出更多差異,絨安菊屬的亞基因組C和D與山絨菊屬以及款冬亞族亞基因組A和B形成一個分支。
透過排除那些在單個基因樹上形成分支但分配給不同亞基因組的款冬亞族序列來過濾數據集後,超矩陣(圖8)和合並方法(補充圖S24;Dryad)產生了相當一致的結果,即也與HomeoSorter共識樹一致。如圖8所示,絨安菊屬的亞基因組D與山絨菊屬形成了一個強支持的前進演化枝(超矩陣支持/局部後驗機率為100%/1.0)。款冬亞族的三個亞基因組分別與短舌菊屬的類群的Caputia菊科/亞基因組A和B(90%/1.0)、千裏光亞科的姐妹組(98%/1.0)和山絨菊屬/絨安菊屬亞基因組D的姐妹組(100%/1.0)分組。特別是,在款冬亞族的三個副本中,絨安菊屬始終是中美洲款冬亞族群的姐妹。具有強大的支持(均為100%/1.0)。
討論
HomeoSorter
盡管仍處於萌芽階段,但將等位基因定相到親本亞基因組的策略對於異源多倍體系統發育非常有利(Oxelman等人2017;Rothfels2021)。除了解決網狀模式之外,它還可以僅使用現有工具對異源多倍體進行下遊分析。然而,大多數當前的等位基因定相方法依賴於簡單的作圖策略。例如,AlleleSorting(Šlenker等人2021)首先根據葉綠體系統發育確定所研究的多倍體的母本,然後根據多倍體等位基因與母本序列的距離將其分配給親本亞基因組。HybPhaser(Nauheimer等人2021)構建了一個初步的系統發育主幹,並將讀取對映到從主幹中選擇的參考文獻。PhyloSD(Sancho等人2022)為二倍體樣本構建了系統發育框架,並透過將多倍體等位基因對映到二倍體框架來對多倍體等位基因進行定相。萊爾等人。(2023)叠代地將多倍體的共有序列與參考序列進行比較,並將位點變異分離到不同的亞基因組中以實作定相。這些作圖策略雖然看似簡約,但可能過於簡化了復雜的異源多倍體後基因前進演化。特別是,他們忽視了ILS的影響, ILS是系統發育不一致的一個眾所周知的原因,由於相似的不一致模式,很難將其與雜交區分開來 (Degnan和Rosenberg2009;Degnan2018),使得結果缺乏說服力。
到目前為止,最復雜的定相同源物模型是AllCoPol(Lautenschlager等人2020)和Homologizer(Freyman等人2023)。AllCoPol是Oberprieler等人(2017)方法的擴充套件,該方法使用排列策略和物種樹重建方法將同源物分配給親本亞基因組(更多詳細資訊,請參閱簡介)。Lautenschlager等人(2020)透過采用啟發式演算法提高其效率。Homologizer是基於AlloppNET(Jones等人2013;Jones2017)開發的,這是一種貝葉斯方法,依賴於多物種合並的參數化模型和馬可夫鏈蒙特卡羅演算法來對跨位點的等位基因進行定相。Freyman等人(2023)透過適應高於四倍體的倍性水平、提供統計節點支持以及記錄下遊分析的等位基因分配來改進它。然而,AllCoPol和Homologizer都受到計算效率的限制。無論我們如何調整參數,AllCoPol都無法在具有16核IntelXeonE5-4657Lv2CPU(2.40GHz)和256GRAM的機器上執行超過180個基因(每個基因一棵樹)的數據集。至於Homologizer,貝葉斯方法的一個常見問題是,對於大型數據集,分析的收斂似乎很難達到。在我們的例子中,即使對於只有100個基因的數據集,Tracerv.1.6(Rambaut等人2014)中估計的主要參數的有效樣本量(ESS)在執行一個多月後通常小於30(約38000幾代人)。因此我們開發了HomeoSorter。透過采用分層策略,HomeoSorter在很大程度上克服了Oberprieler等人方法的擴充套件問題。(2017),計算時間總是與基因數量線性相依(圖6f)。基於模擬測試和我們對款冬亞族的經驗案例,我們進一步證明 HomeoSorter是多倍體系統發育學的有用且可靠的工具。 下面我們更詳細地討論它的優點和註意事項。
HomeoSorter的驗證。 在大多數模擬測試中,HomeoSorter都做得非常好,成功地恢復了自體/異源多倍體模式並準確地對等位基因定相(圖5)。相比之下,AllCoPol在同源多倍體推斷方面比HomeoSorter的效能稍好,能夠在ILS較嚴重的情況下恢復同源多倍體模式(CTs≥0.5vs.CTs≥4;補充圖S18f;Dryad)。然而,它在等位基因分配方面的準確性要低得多,即使在沒有不平等親本貢獻和嚴重ILS問題的情況下,準確性也僅為77%左右(相對於接近100%)(補充圖S18a-c;Dryad)。MPL模型在親本貢獻極其不平等(例如,10:90)的異源多倍體病例上優於HomeoSorter(補充圖S19i,j;Dryad),但在ILS問題更嚴重的病例上表現較差(例如,失敗)當CT≤6與CT≤2時;圖5f,補充圖S19k,l;MPAllopp在研究預先指定的多倍體時,在親本貢獻嚴重不平等的異源多倍體情況下也優於HomeoSorter(補充圖S20h,j;Dryad)。然而,當未指定所研究的多倍體時,MPAllopp分析通常會失敗(補充圖S20a、c、e、g、i、k;Dryad)。此外,MPAllopp無法處理復雜的情況,例如我們的Phylonet_A1和A16數據集。這有點令人驚訝,因為它通常在簡單情況下執行得非常快,但經過我們的反復測試證實了這一點。最重要的是,MPL和MPAllopp都沒有實作定相等位基因的功能。
對於我們的款冬亞族經驗案例,HomeoSorter似乎也正確解析了其異源多倍體模式。首先,基於改組(圖7)和引導(補充圖S21;Dryad)分析的HomeoSorter共識樹與Phylonet分析的結果一致(圖2、3)。其次,基於排序和過濾同源的共識樹(圖8,補充圖S24;Dryad)也與HomeoSorter共識樹(圖7)幾乎相同,並且具有更高的節點支持。第三,推斷的模式得到了基本染色體數量的重要證據的有力支持(見下文)。所有這些結果有力地支持了HomeoSorter的高精度和可靠性。
HomeoSorter的註意事項。 —但是,在某些具有挑戰性的情況下使用HomeoSorter時也應小心謹慎。首先,當親本貢獻極其不平等時,HomeoSorter可能無法辨識異源多倍體模式。例如,在親本貢獻為10:90的情況下(圖5e),即使沒有嚴重的ILS問題,HomeoSorter也幾乎將異源多倍體誤認為同源多倍體,兩個亞基因組始終被推斷為親本的連續姐妹(90%)貢獻(CT在8-200之間;圖5e)。這可能是因為ASTRAL程式傾向於支持等位基因數量更平衡的亞基因組組合。許多本應屬於同一亞基因組的等位基因被分配到不同的亞基因組,導致兩個亞基因組都被單獨的親本吸引。
其次,HomeoSorter的能力在一定程度上受到ILS問題的限制。HomeoSorter未能恢復所有CT≤2的同源多倍體病例和CT≤0.5的異源多倍體病例的前進演化模式。等位基因分配的準確性受到的影響更大,由於CT<6,其下降較早(圖5)。然而,應該指出的是,這裏等位基因分配的準確性可能被低估了。在生成合並樹時,HybridSim還會指示等位基因的歸屬,並且這些指示完全被HomeoSorter接受作為評估等位基因分配的標準。對於CT≥100,合並樹都與模擬網路/樹一致,僅分支長度不同,並且等位基因始終是其相應親本的姐妹。在這些情況下,實際上可以透過視覺辨識多倍體模式。然而,隨著ILS變得更加嚴重,不一致的基因樹迅速增加。當CT≤6時,許多等位基因實際上與合並樹上的另一個親本關系更密切,甚至形成一個分支。在這些情況下,HomeoSorter根據遺傳距離分配等位基因是合理的,但結果肯定違反了HybridSim的指示,使得分配顯得不太準確。
隨機性是可能影響等位基因分配的另一個問題。 這是AllCoPol(Lautenschlager等人2020)和Homologizer(Freyman等人2023)共有的常見問題。對於HomeoSorter來說,主要有三個原因。首先,這是由於HomeoSorter的分層策略,當等位基因與其他基因分組時,在每個子步驟中都會重新評估等位基因。即使等位基因最初被正確分配,這也可能改變分配。其次,盡管在測試了許多程式之後,ASTRAL程式在靈敏度、計算效率和統計一致性方面是HomeoSorter的最佳選擇,但它們仍然經常無法辨別競爭等位基因/亞基因組組合之間的細微差異,並產生相同的最終歸一化四重奏分數。在這種情況下,HomeoSorter將進行隨機選擇,從而產生隨機等位基因分配。第三,復雜的前進演化模式使情況變得更加復雜。例如,當在第一輪ASTRAL分析中單獨分析絨安菊屬時,其兩個亞基因組(C和D)與山絨菊屬的關系為((絨安菊屬_亞基因組_D,山絨菊屬),絨安菊屬_亞基因組_C)。當亞基因組同時存在時,很容易區分它們,但是,如果其中一個亞基因組不存在,HomeoSorter就無法區分它們,只能進行隨機等位基因分配。這可能是我們基於已排序但未過濾的同源物的結果與Phylonet和HomeoSorter分析不一致的主要原因。絨安菊屬亞基因組D的一定數量的等位基因可能被錯誤地分配給亞基因組C,導致亞基因組C被山絨菊屬吸引(補充圖S23;Dryad),而不是巢狀在款冬亞族的亞基因組C中。(圖7、8,補充圖S24;Dryad)。
附加說明 :HomeoSorter的三個重要參數(「--allele」、「-best」和「--every」)值得附加說明。「--allele」(等位基因的數量,作為將基因分成小組的標準)和「--every」(匯集在一起的基因組的數量)參數直接控制要生成的等位基因/亞基因組組合並在第一次ASTRAL分析中計算,「--best」指定在第一次ASTRAL分析中選擇並傳遞到第二次ASTRAL分析的頂級等位基因/亞基因組組合的數量。這些參數的值越大,表明搜尋越詳盡,有助於準確恢復前進演化模式,但會增加計算時間(圖6a-c),並對具有許多研究的多倍體和/或多倍體的復雜情況造成特別沈重的負擔。高倍性水平(圖6d,e)。因此,最好在正式分析之前調整這些參數,以在合理的計算時間和潛在的更高準確度之間取得平衡。特別值得註意的是,如果「--best」設定為「1」,HomeoSorter實際上會跳過第二個ASTRAL分析,單獨分析多倍體,僅合並最後一步的結果。這正是Oberprieler等人采取的策略。(2017)和Lautenschlager等人。(2020)。然而,對於密切相關的多倍體,我們強烈建議進行第二輪ASTRAL分析。在我們的例子中,這會產生更穩定的結果。
款冬亞族的異源多倍體
透過深入的研究,我們揭示了款冬亞族的復雜起源。它包含兩大類群:款冬亞族和類款冬亞族組,具有連續的異源多倍體起源。兩次異源多倍體事件首先產生了現存的款冬亞族。然後,經過早期的多樣化,款冬亞族的一個主要群體。參與第三次異源多倍體事件並產生類款冬亞族組。下面我們將更詳細地討論這些發現。
異源多倍體事件的數量。 ——根據所有證據,總共三個網狀事件將是對款冬亞族起源最有利的情況。和類款冬亞族組。由於當添加更多網狀結構時,Phylonet分析幾乎總是會生成更高的對數偽似然性,因此直接確定網狀結構的真實數量並不容易(Cao等人2019)。曹等人提出的實用策略。(2019)是觀察所得的對數偽似然;當該值開始達到穩定水平時,相應的場景將是「正確的」場景。盡管沒有在所有條件下進行充分測試(Cao等人2019),但這種策略在我們的案例中非常有意義。對於Phylonet_A1(圖2d)和A16(圖3c),當款冬亞族出現相同的三個網狀事件時(註意這是Phylonet_A1的四事件測試,其中包括疆千裏光屬的額外網狀),對數偽-可能性也開始達到穩定水平(圖2g、3g)。後來使用更多網狀結構的測試僅略微提高了該值(圖2g、3g)。更重要的是,這三個網狀事件自從第一次出現以來就一直被推斷出來,而所有額外的網狀結構都與款冬亞族的起源無關。和類款冬亞族組(圖2、3)。
基礎染色體數和HomeoSorter分析的證據進一步支持了這種模式。染色體證據將在下面討論。對於HomeoSorter分析(圖7),以及基於排序和過濾同源物的系統發育樹(圖8,補充圖S24),對款冬亞族的三個亞基因組進行了排序。並分別分為三個不同的譜系。至於類款冬亞族組,有四個亞基因組,但它們只與兩個祖先相關,其中一個亞基因組與親本譜系為姐妹,其他三個亞基因組分別與第二親本譜系的三個亞基因組聚類(圖7、8,補充)圖S24)。三個網狀事件也是這種系統發育模式最簡約的要求。
款冬亞族的異源多倍體 ——我們揭示了現存款冬亞族多樣化之前的兩個異源多倍體事件。第一個發生在千裏光亞族的祖先和與短舌菊屬聯盟有關的譜系之間。然後所得的雜交譜系與山絨菊屬的祖先雜交並產生款冬亞族。與千裏光亞科和山絨菊屬相關的親本譜系非常清晰,正如Phylonet(圖2、3)和HomeoSorter(圖7、8)分析生成的網路和樹上明確且一致地證明的那樣。然而,與短舌菊屬的類群相關的一個需要一些註釋。短舌菊屬的類群本身的特征還在於具有x=30的高堿基染色體數(Robinson等人,1997年;Nordenstam,2007年)。在單個基因樹上,很容易觀察到該聯盟的多個基因拷貝,其中一個或兩個與Caputia密切相關,而其他則距離較遠且位置可變,表明潛在的異源多倍體起源。我們的Phylonet分析(Phylonet_A1和A16)最初沒有檢測到該聯盟的網狀訊號,Caputia是它的姐妹,也是款冬亞族的親本譜系。直接連線到它(圖2、3)。當預先指定為雜種(Phylonet_A17)時,短舌菊屬的類群確實透過網狀事件推斷出來(圖4),兩個親本譜系都與Caputia最密切相關,並且其中一個親本譜系與款冬亞族共享。(圖4c-f)。在預先指定下,這顯然是短舌菊屬的類群的主導模式,因為不同測試之間的結果非常一致(圖4c-f),並且對數偽似然值幾乎相同(圖4g)。然而,由於預先指定,實際上很難確定這是真實的模式還是人為的模式,因為考慮到短舌菊屬的類群和Caputia之間的關系,建議的模式與Phylonet_A1的結果沒有本質上的不同(圖2)和A16(圖3)。此外,考慮到許多個體基因樹上存在與Caputia遠親相關的不同基因拷貝,網狀模式可能不足以代表聯盟的潛在網狀歷史。相反,HomeoSorter分析可以更好地總結潛在的異源多倍體模式。在這三個分選的亞基因組中,兩個與Caputia和款冬亞族的亞基因組A歸為一類,另一個則關系較遠,但位置尚未確定(圖7)。
與短舌菊屬屬密切相關的Caputia屬使解開短舌菊屬聯盟起源的困難變得更加復雜。盡管具有x=10的低堿基染色體數,但基於葉綠體(cp)和核糖體(nr)基因樹上的不一致位置,Caputia也被認為是一個雜種屬,其中Caputia巢狀在千裏光亞科內並且是姐妹分別屬於短舌菊屬的類群和千裏光亞科遠親(Pelser等人2010)。這一假設得到了Caputia中間花微形態學的進一步支持(Nordenstam2012),這是千裏光亞族分類的關鍵(Nordenstam等人2009)。Caputia在nrDNA樹上的位置(Pelser等人2010)由我們的SLCN基因證實(圖2、3、7、8)。然而,僅憑SLCN基因,實際上並沒有明確的線索表明Caputia的雜交起源。在用於系統發育分析的727個基因中,只有24個基因對Caputia發出了旁系同源警告(C.medley-woodii10個,C.scaposa19個,其中5個是共享的)。此外,即使對於這24個基因,除了4個基因外,Caputia的序列都形成了一個強支持的前進演化枝,表明它們可能是同源等位基因,或者只是在屬分化後重復。此外,Phylonet分析表明該屬沒有雜交訊號(圖2、3)。因此,短舌菊屬聯盟和Caputia的情況要復雜得多,我們無法對它們的前進演化模式提供非常有信心的答案。但對款冬亞族的結果影響並不顯著。款冬亞族之間的密切關系。而短舌菊屬的聯盟是可以保證的,不確定性只集中在款冬亞族是否成立上。與祖先或短聲聯盟的祖先共享親本血統。根據目前的證據,前一種假設更為有利。
類款冬亞族類群的異源多倍體。 類款冬亞族類群的異源多倍體模式非常簡單。基於形態相似性,該類群通常被認為是款冬亞族的核心成員(Bremer1994;Robinson等人1997;Nordenstam2007)。在之前的分子系統發育研究中(Pelser等人2007,2010),它也始終是中美洲款冬亞族群的姐妹。在cpDNA和nrDNA樹上,因此最初沒想到它與其他款冬亞族物種有不同的起源。然而,Phylonet和HomeoSorter分析強烈表明了獨立的異源多倍體起源。Phylonet一致推斷類款冬亞族群體存在網狀事件,其親本與山絨菊屬譜系和中美洲款冬亞族群體有關。(圖2、3)。HomeoSorter對類款冬亞族群的四個亞基因組進行了分類,其中一個是山絨菊屬的姐妹組,另外三個分別是中美洲款冬亞族群的三個亞基因組的姐妹。(圖7、8,補充圖S24;Dryad)。
染色體證據。 重要的是,推斷的款冬亞族異源多倍體模式得到了基本染色體數證據的有力支持。對於款冬亞族,兩個祖先山絨菊屬和千裏光亞科的現存最近親,其基本染色體數x=10(Hunziker等人,1989年;Nordenstam等人,2009年)。短舌菊屬的類群和Caputia與第三個祖先密切相關,其特征是x=30(例如Ornduff等人,1963;Hair1966;Beuzenberg1975)和x=10(Afzelius1967),這表明第三個祖先親本譜系很可能也基於x=10。款冬亞族盡管有一些例外(例如,x=9,在兩棲太平洋組中很少為7;Ornduff1963;Borgmann1964;Schaack等人1974),但主要基於x=30(Nordenstam等人,2009),與此處推斷的異源多倍體模式高度一致。
至於類款冬亞族群體,山絨菊屬代表親本譜系之一,並且已註意到x=10(Hunziker等人,1989)。另一個親本譜系,中美洲款冬亞族群,所有屬都基於x=30(例如,Pippen1968;Turner等人1973;Robinson等人1997;Palomino等人2018),除了x=30以外。Arnoglossum為25(Jones1970),YermoDorn沒有數據。因此,觀察到的類款冬亞族群體的唯一堿基染色體數x=40(Turner等人,1967年;Robinson等人,1997年)與推斷的異源多倍體模式完全匹配。
結論
在這項研究中,我們提出了HomeoSorter,一種用於異源多倍體系統發育研究的新同源分類管道。HomeoSorter克服了當前同源分類工具的縮放問題,並在辨識多倍體模式和分配等位基因方面表現出高精度。它是異源多倍體系統發育學的可靠且急需的工具,特別是在當前的基因組時代。使用HomeoSorter結合Phylonet的MPL模型和基因組規模數據,我們為款冬亞族的起源提供了強有力的證據。我們的分析表明,款冬亞族包含兩大類群: 款冬亞族和類款冬亞族組,具有連續的異源多倍體起源和相互關聯的關系。 特別是,異源多倍體模式得到了基礎染色體數量的重要證據的有力支持。如此清晰且有充分支持的案例在多倍體研究中很少見,並 為未來研究這種現象及其潛在的前進演化效應提供了極好的模型。
圖表
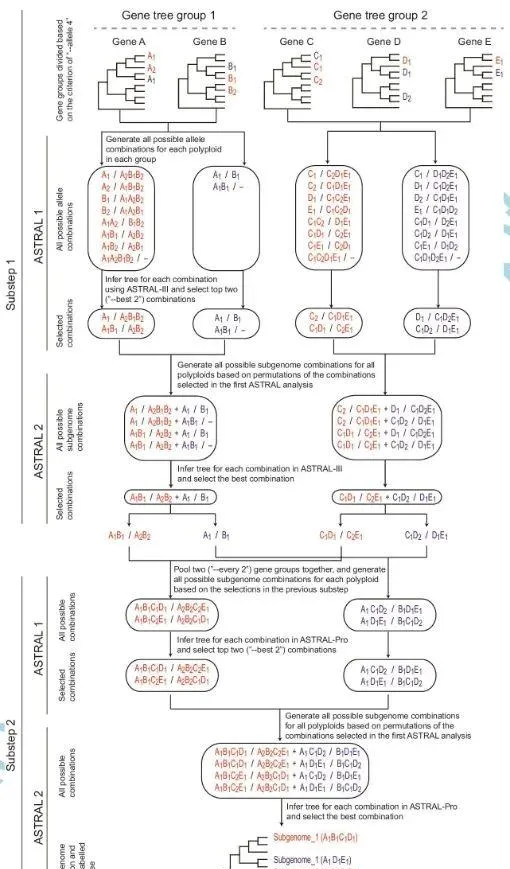
圖1. HomeoSorter核心程式的工作流程,使用具有五個基因(A–E)、兩個四倍體(紅色和藍色)以及參數―--allele4-best2--every2的簡單案例進行說明」。A-E後面的下標表示不同的等位基因。子基因組由斜線(「/」)分隔,破折號(「–」)表示沒有等位基因分配給子基因組。參數「--allele4」表明基因分組時,每組中每個多倍體的等位基因總數不能超過4個。例如,前三個基因不能組合在一起;否則,紅色多倍體的等位基因總數將為六個,超出限制。參數「--best2」表明將在第一次ASTRAL分析中選擇前兩個等位基因/亞基因組組合,並在第二次ASTRAL分析中重新評估,「--every2」表明每兩個基因組將逐漸組合從第二個子步驟開始。請註意,這個範例非常簡單。整個過程僅需要兩個子步驟,並且僅生成一些可能的等位基因/亞基因組組合。對於大型數據集,第二個子步驟需要重復多次,直到最終包含所有基因。此外,在更復雜的情況下(例如更多的多倍體、更高的倍性水平胡/或更大的參數值),HomeoSorter將生成更多數量的等位基因/亞基因組組合。
圖2. 使用最大偽似然(MPL)模型(Phylonet_A1)進行第一次Phylonet分析的結果。六次測試的結果網路分別允許一到六個網狀結構,如(a)-(f)所示。橙色表示千裏光亞族的樣本,藍色表示短舌菊屬類群的樣本,紅色表示款冬亞族的樣本,綠色表示Gynoxyoid組的樣本。網狀節點邊緣附近的數位表示繼承機率。柱形圖(g)說明了六次測試的結果對數偽似然,表明從第四次測試開始,值開始達到穩定水平。
圖3. 使用MPL模型(Phylonet_A16)進行第十六次Phylonet分析的結果。六次測試的結果網路分別允許一到六個網狀結構,如(a)-(f)所示。橙色表示千裏光亞族的樣本,藍色表示短舌菊屬類群的樣本,紅色表示款冬亞族的樣本,綠色表示Gynoxyoid組的樣本。網狀節點邊緣附近的數位表示繼承機率。柱形圖(g)說明了六次測試的結果對數偽似然性,表明從第三次測試開始,值開始達到穩定水平。
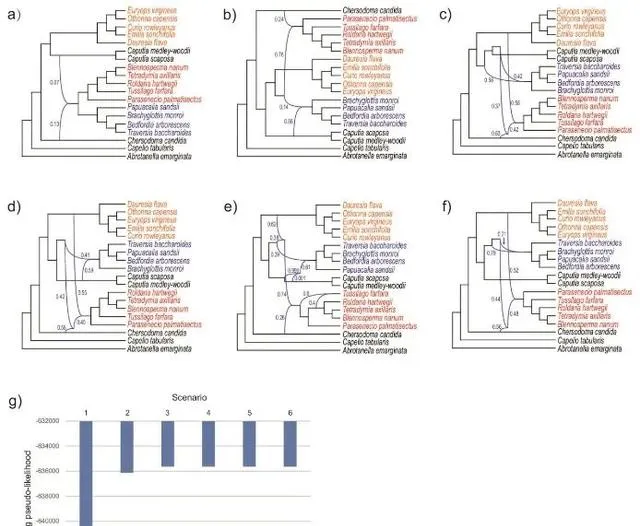
圖4. 使用MPL模型(Phylonet_A17)進行第十七次Phylonet分析的結果。六次測試的結果網路分別允許一到六個網狀結構,如(a)-(f)所示。橙色表示千裏光亞族的樣本,藍色表示短舌菊屬類群的樣本,紅色表示款冬亞族的樣本,綠色表示Gynoxyoid組的樣本。網狀節點邊緣附近的數位表示繼承機率。柱形圖(g)說明了六次測試的結果對數偽似然性,表明從第三次測試開始,值開始達到穩定水平。

圖5. 基於模擬數據的HomeoSorter在不同場景下的測試結果,其中(a)-(e)為不同親本貢獻的異源多倍體,(f)為同源多倍體。折線圖表示正確辨識親本(a-e)或同源多倍體模式(f)的百分比,箱線圖描述了100個改組重復中正確等位基因分配的變化(a-e)。結果表明,HomeoSorter在大多數情況下都表現良好,但在譜系排序嚴重不完整和/或親本貢獻極其不平等的情況下,其效能會下降。
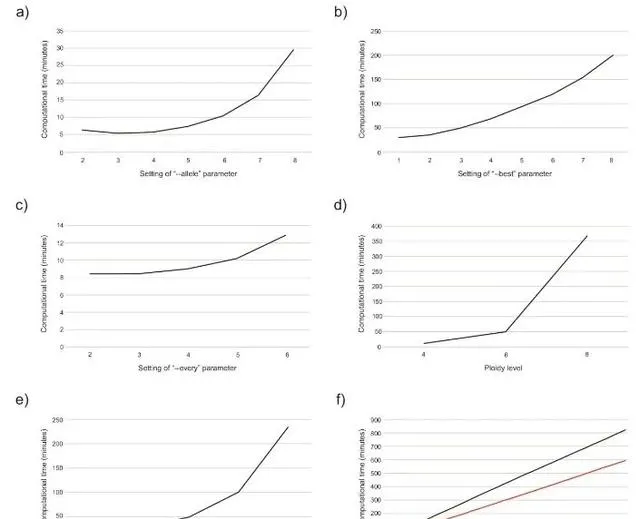
圖6. 計算時間與HomeoSorter分析的六個主要因素之間的相關性。這六個因素包括參數「--等位基因」(a)、「--最佳」(b)、「-每個」(c)、倍性水平(d)、多倍體數(e)和基因編號(f)。除非另有說明,所有測試均使用單個四倍體和HomeoSorter的預設設定執行。對「--best」參數(b)的測試是用三個四倍體進行的。對於基因編號(f),測試了三種不同的場景,藍線表示基於四倍體和參數「--allele4--best1--every2」的結果,紅色表示兩個四倍體,「--allele6--best3--every3」,黑色表示六倍體,「--allele6--best1--every2」。請註意,此處的計算時間是使用單個執行緒評估的。在實際使用中,可以采用多執行緒來節省時間。
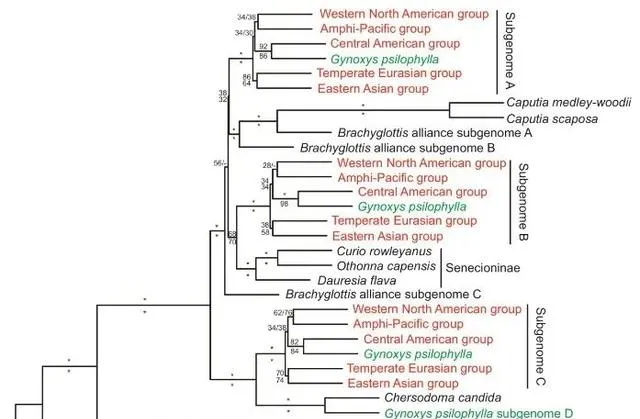
圖7. 基於50次改組重復的HomeoSorter分析的多數共識樹。紅色表示款冬亞族的五個主要類群。綠色代表Gynoxyoid組。基於50個改組和50個引導重復總結的支持值(%)分別顯示在分支的上方和下方,其中星號(「*」)表示完全支持,破折號(「–」)表示與引導分析不同的關系。
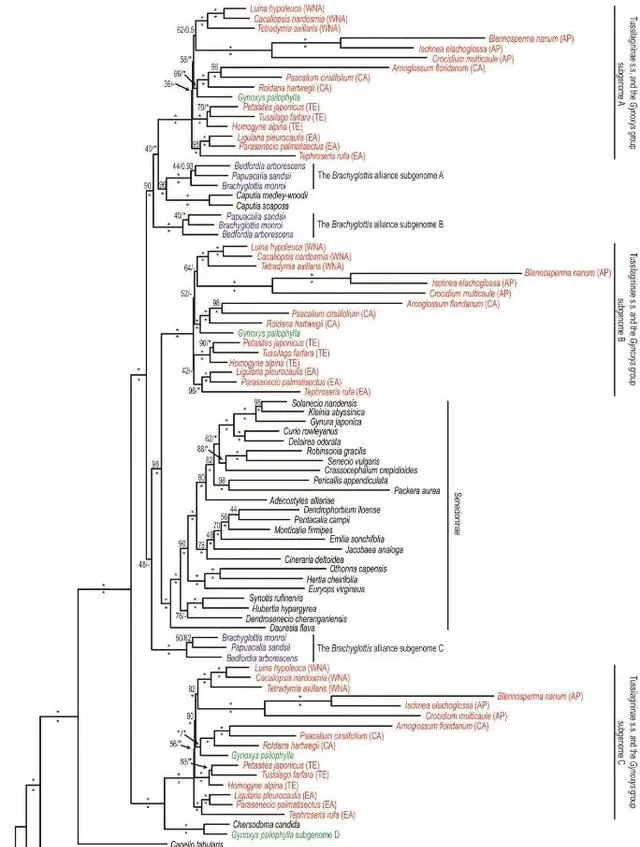
圖8. 從50個超矩陣最大似然樹中總結出的多數共識樹,這些樹是根據排序和過濾的同源物重建的。紅色表示款冬亞族的樣本,綠色表示Gynoxyoid組的樣本,藍色表示短舌菊屬類群的樣本。款冬亞族後面括弧內的縮寫。樣本表示樣本所屬的五個主要類群,「AP」代表兩棲太平洋類群,「CA」代表中美洲類群,「EA」代表東亞類群,「TE」代表溫帶歐亞類群,「WNA」代表北美西部群體。超矩陣方法的支持值(%)和合並方法的局部後驗機率分別顯示在分支的上方和下方,星號(「*」)表示完全支持,破折號(「–」)表明合並方法表明了不同的關系。
期刊:Systematic Biology
文章標題:Complex but Clear Allopolyploid Pattern of Subtribe Tussilagininae (Asteraceae: Senecioneae) Revealed by Robust Phylogenomic Evidence, with Development of a Novel Homeolog-Sorting Pipeline
作者資訊:Chen Ren,Long Wang,Ze-Long Nie,Ming Tang,Gabriel Johnson,Hui-Tong Tan,Nian-He Xia,Jun Wen,Qin-Er Yang
原文連結:https://doi.org/10.1093/sysbio/syae046
文內圖片及封面圖片來源原文











