副标题:有机硼酸酯的立体选择性亚乙烯基同系化反应合成烯基硼酸酯
同系化(homologation)反应在有机合成中具有广泛的应用,能够在不改变原始反应基团的情况下通过链延伸或环扩张直接编辑分子骨架。硼的同系化反应(即Matteson反应)通过精确控制加成顺序和立体化学,在可编程或自动化有机合成中变得愈发重要。如图1a所示,在经典的Matteson反应中卡宾以迭代方式插入硼酸酯的C-B键中,从而将sp3杂化的碳引入分子骨架中。此外,虽然氮杂-和氧杂-Matteson反应已有报道,但将sp2杂化的碳(特别是烯烃单元)插入硼酸酯C-B键中仍极具挑战性。鉴于烯烃在功能有机分子骨架中普遍存在(图1b),因此如何实现立体选择性亚乙烯基插入以产生烯基硼酸酯就变得极为重要。目前,化学家已经报道了许多有效的方法来制备烯基硼酸酯,包括:炔烃硼氢化反应、bora-Wittig反应、Heck反应、交叉复分解等,但是从有机硼酸酯开始的合成研究却很少。尽管使用含硼保护基的双功能构建砌块可以通过钯催化的Suzuki偶联反应实现形式的烯烃插入,但是这种方法需要硼保护和脱保护,并且与烷基硼酸酯的交叉偶联并不是一个简单的反应。此外,Zweifel烯化是将烯基偶联到C-B键上的一种优雅方法(图1c),不过原始硼酸酯基团在反应过程中会被消除。
近日,美国 芝加哥大学董广彬 教授与 匹兹堡大学刘鹏 教授等研究者 通过顺序和非对映选择性插入硅烷基和烷氧基取代的carbenoids,然后进行原位立体特异性消除,便可以良好的产率和优异的 trans -选择性将烷基和芳基硼酸酯转化为亚乙烯基同系物 (图1d) ,从而成功地实现了无保护基或贵金属催化剂的「硼到硼(B-to-B)」转化 。此外,密度泛函理论(DFT)计算揭示了carbenoid插入的非对映选择性起源以及具有不同硫化物结合亲和力的Lewis酸如何影响竞争性SN2-和SN1-型1,2-硼酸盐迁移途径。相关成果发表在 Nature Synthesis 上。
图1. 亚乙烯基同系化及策略设计。图片来源: Nat. Synth.
首先,作者选择烷基频哪醇硼酸酯 Int-1a 为模板底物对反应条件进行筛选(图2),并获得最佳反应条件:即 Int-1a 先与大位阻锂化烷氧基芳基硫烷试剂 S-1 (氧取代carbenoids前体)在-78℃反应得到ate-络合物中间体,再在Lewis酸(ZnCl2)的作用下进行1,2-金属盐迁移并将氧取代的亚甲基插入C-B键中,接着在室温下用1.2 equiv. pSO4对所得的α-烷氧基-β-甲硅烷基硼酸酯中间体进行简单的一锅法处理,便可以91%的产率和13:1的 E / Z 选择性得到所需的烯基硼酸酯产物 2l 。此外,当在添加ZnCl2之前将反应混合物温热至室温时,反应产率较高(90%),但 E / Z 选择性较低(4:1)。
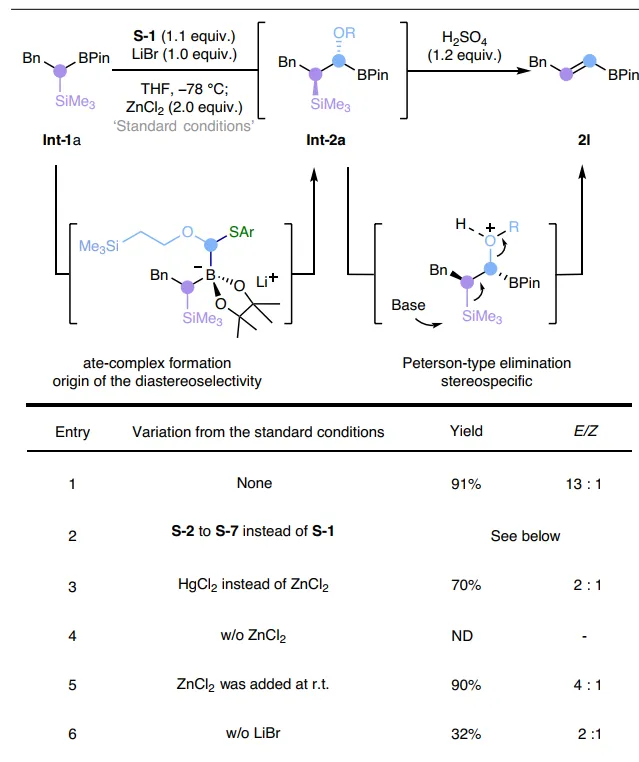
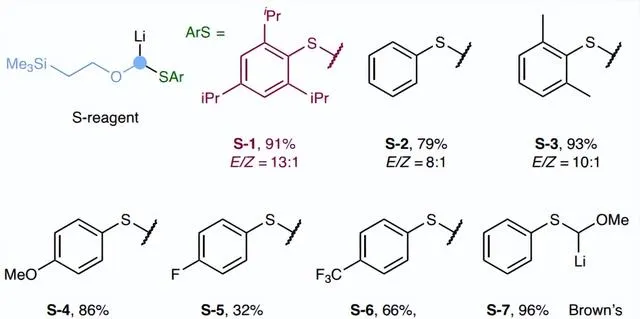
图2. 反应条件优化。图片来源: Nat. Synth.
在最优条件下,作者对烷基硼酸酯的底物范围进行了考察(图3),结果显示多种伯烷基硼酸酯均能兼容该反应,以良好的产率和中等至优异的 E / Z 选择性(高达19:1)获得相应的同系化产物,同时还能够耐受多种官能团,包括:芳基氟化物( 2b、2i )、芳基氯化物( 2c、2j、2k )、芳基溴化物( 2d )、硫醚( 2h )等。当仲烷基硼酸酯在标准条件下进行反应时仅以较低的产率获得所需的 E -烯烃产物,这可能是由于空间位阻增加所致。为此,作者通过使用新戊二醇衍生的硼酸酯(Bneop)来减少硼周围的空间位阻,从而提高反应活性;同时使用位阻更大的二甲基苯基硅烷基衍生的carbenoid提供了更好的非对映选择性,并且HgCl2的使用在一定程度上进一步提高了 E / Z 选择性。为了简化分离过程,作者将烯基Bneop产物直接转化为相应的烯基碘化物。类似地,不同环尺寸的环烷基硼酸酯( 3p-3s )、带有桥环骨架底物( 3t 、 3u )、链状仲烷基硼酸酯( 3w )甚至复杂天然产物衍生的底物( 3v 、 3x )均能有效地实现这一转化并获得所需产物。
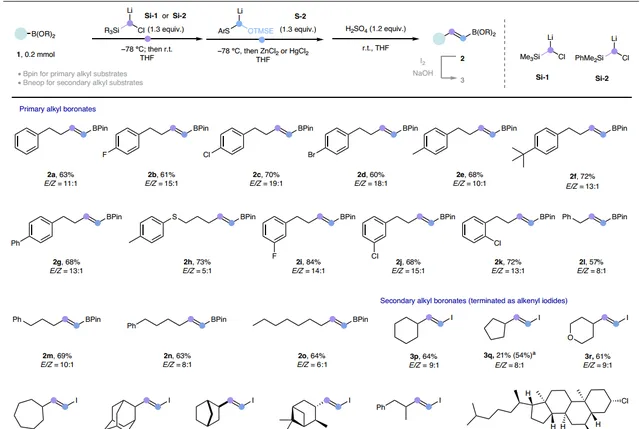
图3. 烷基硼酸酯的底物范围。图片来源: Nat. Synth.
接下来,作者使用HgCl2作为Lewis酸对芳基硼酸酯的底物适用性进行了研究(图4),结果显示无论芳环对位、邻位或间位带有吸/供电子基团(如:三氟甲基醚( 5b )、硫醚( 5c )、硅烷基醚( 5d )、碘化物( 5e )、硅烷( 5f )、烷基氯( 5h )、烯基( 5i )、炔基( 5j )等),都能以良好的产率(45-76%)和优异的 E / Z 选择性(>20:1)获得相应产物( 5a-5t ),特别是产物 5a 能以5 mmol规模进行制备(总产率:50%)。此外,杂芳烃衍生的硼酸酯( 5u-5y )和雌酮衍生的底物( 5z )也能顺利地实现这一转化并得到同系化产物。
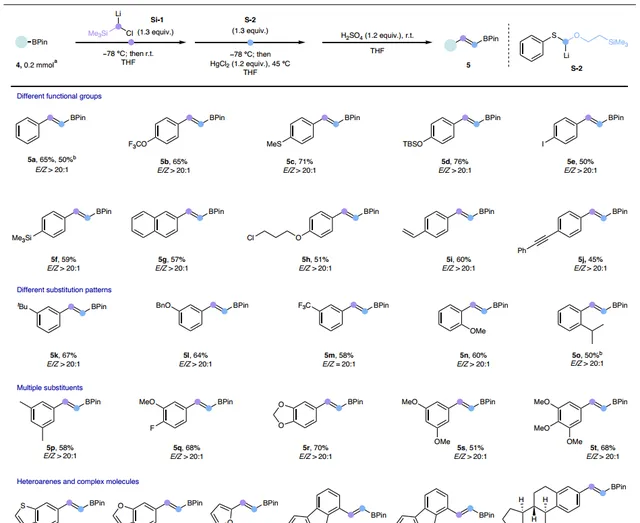
图4. 芳基硼酸酯的底物范围。图片来源: Nat. Synth.
为了进一步探究反应机理,作者成功分离出α-甲氧基-β-硅烷基硼酸酯中间体( anti - 7a )并对其进行表征(图5a),该中间体用酸处理仅产生 E -产物 5a ,进而证实了其在Peterson型消除步骤中的中间作用。为了进一步确定相对立体化学,作者将α-甲氧基-β-硅烷基硼酸酯中间体( anti - 7b )进行亚甲基同系化反应,然后经氧化和p-nosyl保护便可得到亚磺酸酯 8 ,其X-射线衍射分析证实硅基和甲氧基处于 anti -构型。其次,作者还通过DFT计算来研究carbenoid插入步骤的非对映选择性和Lewis酸效应。如图5b所示,烷氧基取代碳负离子 11 与α-甲硅烷基硼酸酯 10 加成过程的计算表明经过渡态 TS1a 形成的硼酸酯 12a 是动力学上有利的产物;而另一个非对映异构体 12b 则由于芳基硫醇盐LG和硼酸酯 10 上α-甲基之间的不利相互作用导致过渡态 TS1b 能垒比 TS1a 高1.3 kcal mol-1。随后,作者探索了ZnCl2的Lewis酸效应(图5c),其中ZnCl2倾向于与芳硫基化物的硫原子和硼酸酯的氧原子结合并形成 13a 。从 13a 开始,经协同过渡态 TS2 进行的SN2型途径是最有利的,活化自由能(Δ G ‡)为15.6 kcal mol-1;而SN1型途径涉及逐步LG解离形成 14 (吸热),然后通过 TS3a 或 TS3b 进行1,2-迁移,因此不太有利。由于立体特异性SN2型1,2-迁移( TS2 )会导致烷氧基取代碳中心的立体化学完全反转,因此该过程将 13a 转化为α-甲氧基-β-硅烷基硼酸酯 15 的 anti -非对映异构体,再在立体特异性Peterson消除后选择性地形成 E -烯烃,这与实验观察到的立体选择性相一致。鉴于汞比锌具有更强的硫醇盐结合亲和力,因此当使用HgCl2作为Lewis酸时,非立体特异性SN1型1,2-迁移途径可能更有利。事实上,HgCl2促进芳基硫醇盐解离的Δ G ‡仅为3.3 kcal mol-1,比类似ZnCl2介导LG解离过程的能垒低12.2 kcal mol-1(图5d)。另外,由于中间体 14 经 TS3a 和 TS3b 进行1,2-迁移的能垒差较小,因此使用更亲硫的Lewis酸HgCl2时逐步SN1型途径更有利,从而导致同系化产物的非对映选择性降低。

图5. 机理研究。图片来源: Nat. Synth.
此外,作者探索了同系化反应的合成应用。具体而言:1)该方法可用于生成掩蔽的烯烃单元,可以耐受氢化条件(图6a);2)当使用4.0 equiv ZnCl2时,无需汞盐即可实现芳基硼酸酯的同系化反应(图6b),并获得单一的 E -异构体;3)该方法的合成潜力在piperdardine家族天然产物的简化合成中得到了进一步证明(图6c)。从共同底物芳基硼酸酯 4r 出发,通过一锅法双亚甲基同系化、亚乙烯基同系化和Suzuki反应便可以三步、32%的总产率、12:1 E / Z 选择性合成piperdardine(Route A),并且该过程仅需一次纯化。类似地,作者通过Route B制备piperdardine类似物,总产率为67%。为了制备三烯天然产物retrofractamide A,作者通过亚乙烯基同系化、双亚甲基插入、亚乙烯基同系化和Suzuki反应实现其合成(Route C)。与先前的方法相比,这种迭代合成策略步骤更少、总产率更高,最大限度地减少反应中间体的纯化,更重要的是为合成设计提供了可编程性。
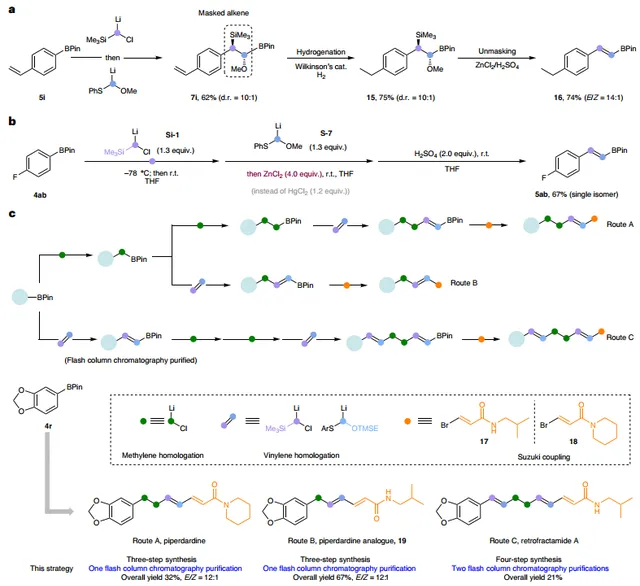
图6. 合成应用。图片来源: Nat. Synth.
鉴于ate-络合物(如: 9a )的形成是非对映选择性的,并且软Lewis酸(如:ZnCl2和HgCl2)在1,2-金属盐迁移过程中促进芳基硫醇盐作为LG,因此用亲氧(即「硬」)Lewis酸(如:AlCl3或CeCl3)代替软Lewis酸就可以将烷氧基作为LG,从而选择性地获得中间体 syn - 20a (>15:1 Z / E ,图7a),比α-甲氧基硼酸酯 7a 具有更强的稳定性,可进行快速柱色谱纯化和光谱表征。为了有效地促进 anti -消除,作者发现对位氯取代的芳基硫醇盐( S-8 )效果更好。当用1.2 equiv TBAF和1.5 equiv甲基乙烯基砜处理中间体 20 时,能以良好的产率和中等的非对映选择性获得所需的 Z -烯基硼酸酯( 21a-f ),其中非对映选择性的降低可能是由于消除步骤中芳基硫醇盐LG引起的差向异构化所致。
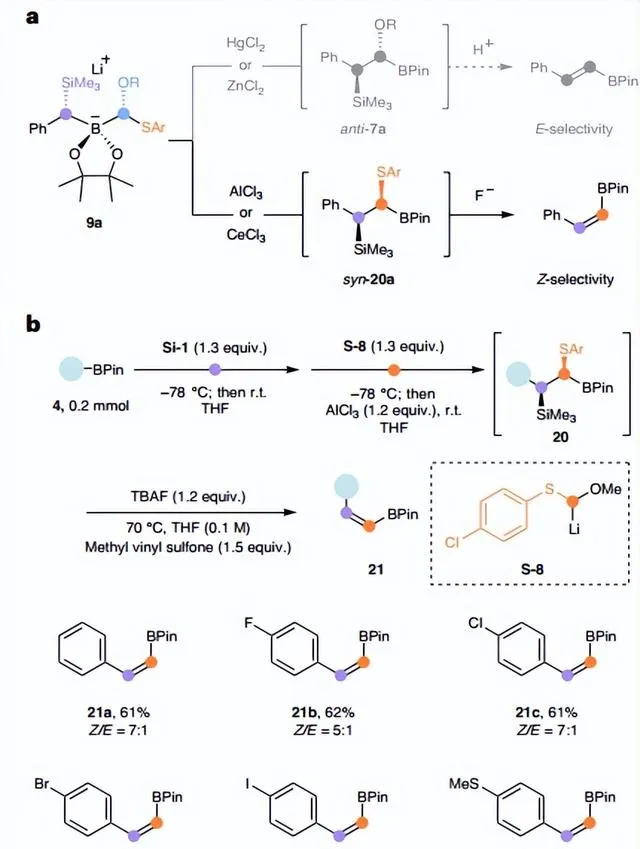
图7. 芳基硼酸酯的 Z -亚乙烯基同系化。图片来源: Nat. Synth.
总结
董广彬课题组和刘鹏课题组通过顺序和非对映选择性插入硅烷基和烷氧基取代的carbenoids,然后进行原位立体特异性消除,便可以良好的产率和优异的 trans -选择性将烷基和芳基硼酸酯转化为亚乙烯基同系物,甚至可以通过迭代亚乙烯基和亚甲基同系化反应来实现piperdardine家族天然产物的可编程合成。此外,DFT计算揭示了carbenoid插入的非对映选择性起源以及具有不同硫化物结合亲和力的Lewis酸如何影响竞争性SN2-和SN1-型1,2-硼酸盐迁移途径。
Synthesis of alkenyl boronates through stereoselective vinylene homologation of organoboronates
Miao Chen , Thomas H. Tugwell, Peng Liu, Guangbin Dong
Nat. Synth ., 2024 , DOI: 10.1038/s44160-023-00461-w











