導讀
近日,南韓科學技術院Mu-Hyun Baik與美國埃默裏大學Simon B. Blakey課題組報道了一種平面手性銠茚基催化劑促進非活化末端烯烴的對映選擇性氮雜環丙烷化反應(aziridination),合成了一系列對映體富集的高價值手性氮雜環丙烷衍生物(aziridines)。同時,該策略具有廣泛的受質範圍、良好的官能團相容性以及優異的化學選擇性等特點。此外,計算研究揭示了一種逐步的氮雜環丙烷化機理,其中烯烴遷移插入起著核心作用。該過程導致形成有張力的四元金屬環,並充當整個反應中的對映選擇性和速率決定步驟。文章連結DOI:10.1021/jacs.3c10637
(圖片來源: J. Am. Chem. Soc. )
正文
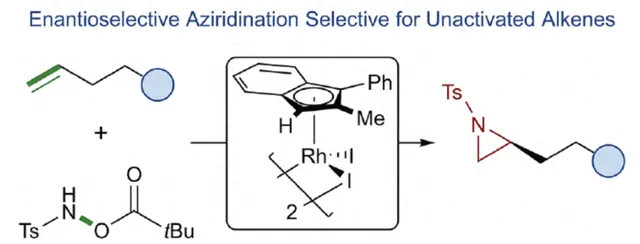
氮雜環丙烷是一種具有價值的張力三元含氮雜環。其立體選擇性合成可以透過三種不同的斷裂方式來實作:(1)手性鹵代胺或胺基醇的分子內縮合,(2)碳源到亞胺的立體選擇性加成,(3)氮賓等價物與烯烴的立體選擇性加成(最具吸重力)。目前,化學家們還開發了大量透過過渡金屬催化立體選擇性合成手性氮雜環丙烷的方法。然而,這些方法集中於活化烯烴(如苯乙烯和 α,β -不飽和體系)的氮雜環丙烷化反應(Scheme 1a,top)。同時,具有烷基取代的非活化烯烴的立體選擇性氮雜環丙烷化反應仍然具有挑戰(Scheme 1a,bottom)。目前,僅只有四種方法實作了非活化烯烴的分子間對映選擇性氮雜環丙烷化反應( Chem. Commun. 2009 , 4266; Chem. Commun. 2012 , 48 , 7188; J. Am. Chem. Soc. 2022 , 144 , 17156; J. Am. Chem. Soc. 2023 , 145 , 7516.)。然而,上述的方法對於非活化受質通常具有較低的產率和對映選擇性。2019年,Rovis課題組( J. Am. Chem. Soc. 2019 , 141 , 12536.)開發了一種Rh(III)-催化非活化末端烯烴的形式[4+1]環加成反應,合成了一系列吡咯烷的方法(Scheme 1b,top)。2020年,Blakey課題組( J. Am. Chem. Soc. 2020 , 142 , 13996.)設計一種用於區域與對映選擇性烯丙基C-H胺化的平面手性銠茚基催化劑(Scheme 1b,bottom)。受此啟發,南韓科學技術院Mu-Hyun Baik與美國埃默裏大學Simon B. Blakey課題組報道了一種平面手性銠茚基催化劑促進非活化末端烯烴的對映選擇性氮雜環丙烷化反應,合成了一系列對映體富集的手性氮雜環丙烷衍生物(Scheme 1c)。 下載化學加APP到你手機,更加方便,更多收獲。
(圖片來源: J. Am. Chem. Soc. )
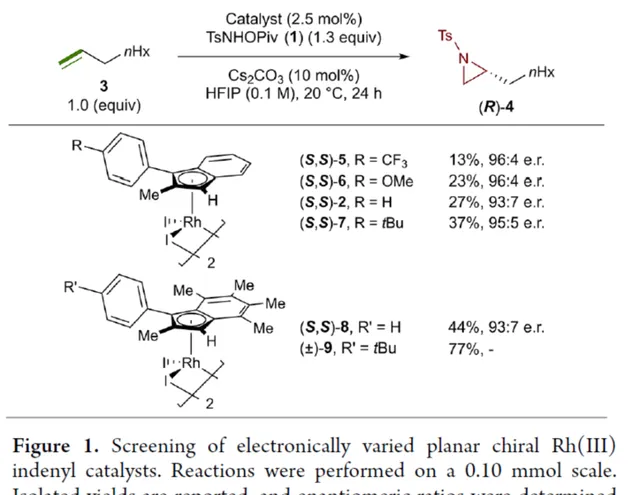
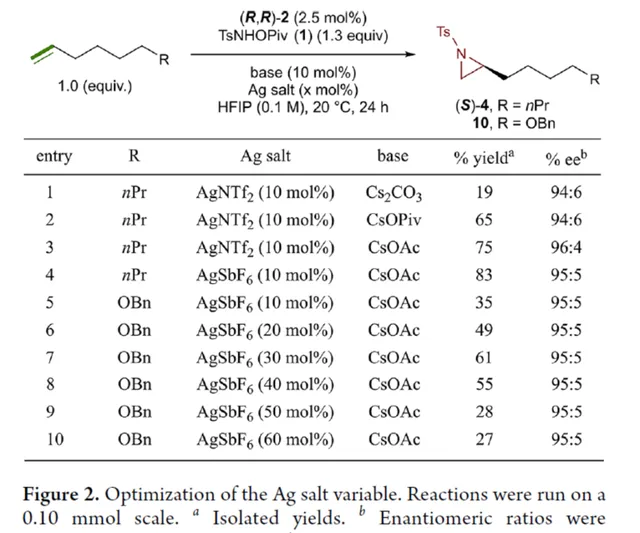
首先,作者對平面手性Rh(III)茚基催化劑(Figure 1)以及反應的條件進行了篩選(Figure 2)。篩選結果表明,當以1-壬烯作為受質,TsNHOPiv( 1 ,1.3 equiv)作為氮源, ( R,R )-2 (2.5 mol %)作為催化劑,AgSbF6(10 mol %)作為銀鹽,CsOAc(10 mol %)作為堿,在HFIP溶劑中20 oC反應24 h,可以83%的收率得到產物 ( S )-4,e.r.為95:5。同時,當以芐基保護的5-己烯-1-醇為受質時,需將銀鹽的負載量提高至3 0 mol %,可以61%的收率得到產物 10,e.r.為95:5。
(圖片來源: J. Am. Chem. Soc. )
(圖片來源: J. Am. Chem. Soc. )
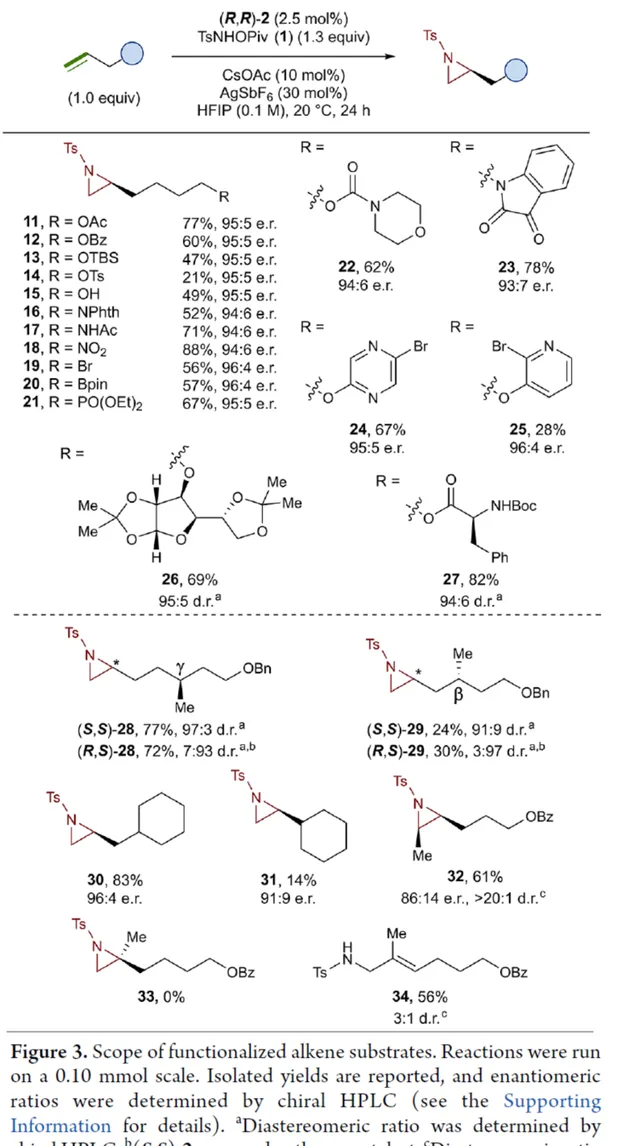
在獲得上述最佳反應條件後,作者對受質範圍進行了擴充套件(Figure 3)。首先,一系列不同取代的末端烯烴受質,均可順利進行反應,獲得相應的產物 11 - 21 ,收率為21-88%,e.r.為93:7-96:4。其中,烷基鹵化物、硼酸酯和膦酸酯等活性的官能團,均與體系相容。含有更為復雜取代的受質(如嗎啉胺基甲酸酯、靛藍、二嗪和吡啶),均為合適的受質,獲得相應的產物 22 - 25 ,收率為28-78%,e.r.為93:7-96:4。受保護的 D -呋喃葡萄糖和 L -苯丙胺酸衍生的烯烴受質,也與體系相容,獲得相應的產物 26 (收率為69%,d.r.為95:5)和 27 (收率為82%,d.r.為94:6)。其次, L -香茅醇衍生的烯烴,在使用兩種不同構型的催化劑時,可分別獲得相應的產物 ( S,S )-28 (收率為77%,d.r.為97:3)和 ( R,S )-28 (收率為72%,d.r.為7:93)。同時,將手性中心轉移至 β -位時,可分別獲得相應的產物 ( S,S )-29 (收率為24%,d.r.為91:9)和 ( R,S )-29 (收率為30%,d.r.為3:97)。烯丙基環己烷和乙烯基環己烷,也是合適的受質,分別獲得相應的產物 30 (收率為83%,e.r.為96:4)和 31 (收率為14%,e.r.為91:9)。此外, Z -二取代的烯烴,可順利進行反應,獲得相應的產物 32 (收率為41%,e.r.為86:14,d.r.> 20:1)。然而,1,1-二取代烯烴未能有效的進行反應(如 33 ),但可獲得末端烯丙基胺產物 34 (收率為56%,d.r.為 3:1)。
(圖片來源: J. Am. Chem. Soc. )
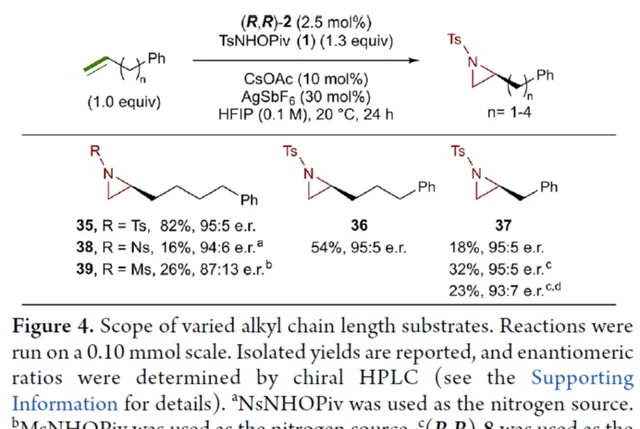
緊接著,作者研究了烷基鏈長度對反應的影響(Figure 4)。研究發現,碳鏈長度為4時不會影響反應(如 35 ),但將碳鏈長度降為3或1時,收率明顯下降(如 36 和 37 )。值得註意的是,在碳鏈長度發生變化時,對映選擇性不受影響。同時,當使用NsNHOPiv與MsNHOPiv作為氮源時,反應的收率也出現明顯的下降,對映選擇性略有降低,如 38 和 39 。
(圖片來源: J. Am. Chem. Soc. )
接下來,作者對同時含有活化與非活化的烯烴受質進行了競爭性反應,用於研究烯烴的電性對於反應的影響(Figure 5)。研究發現,氮雜環丙烷化僅發生在非活化的烯烴上,而活化的烯烴保持完整,從而表明在對映選擇性氮雜環丙烷化方法中具有全新的選擇性水平。

(圖片來源: J. Am. Chem. Soc. )
此外,作者提出了兩種合理的催化迴圈過程(Scheme 2)。第一種機理涉及醯胺的形成以及隨後的烯烴插入過程(黑色)。第二種機理涉及氮賓形成(藍色和綠色)。由於其不切實際的高能量需求,從而排除了烯烴直接進行協同金屬化-去質子化(CMD)的可能性。

(圖片來源: J. Am. Chem. Soc. )
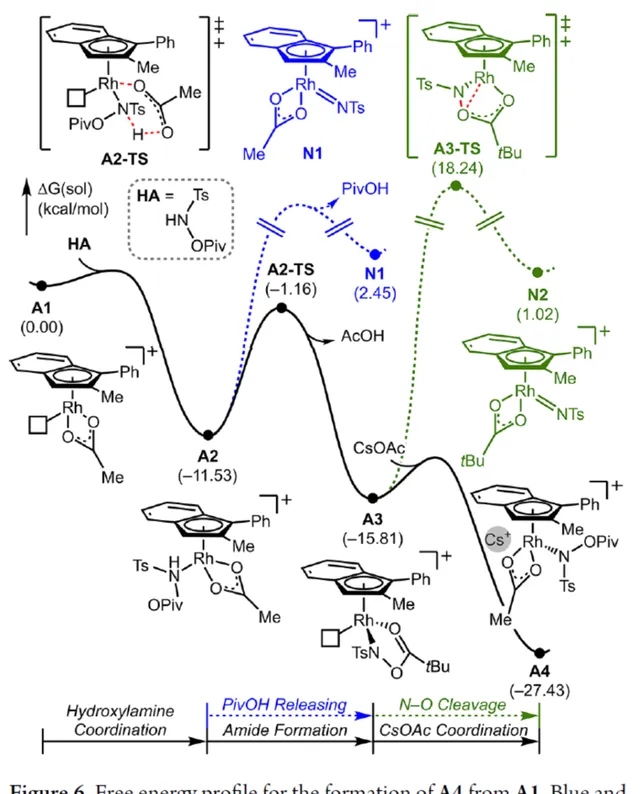
為了進一步證明上述機理的合理性,作者進行了相關的理論計算研究(Figures 6-8)。首先,羥胺HA與 A1 的配位生成18-電子的配合物 A2 。配合物 A2 經分子內去質子化,生成金屬-醯胺配合物 A3 ,並釋放乙酸。配合物 A3 與堿基結合,生成銫-結合配合物 A4 。值得註意的是,在 A4 的形成過程中,兩個中間體 A2 和 A3 有可能形成氮賓中間體 N1 和 N2 。然而,由於其所需的高能量表明反應不是透過氮賓中間體進行的。由於 A4 是飽和狀態,它釋放CsOAc並生成16-電子配合物 A3 (Figure 6)。
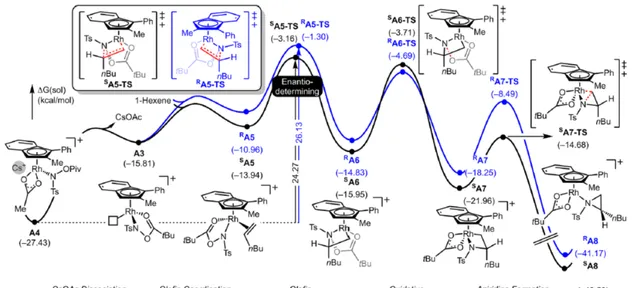
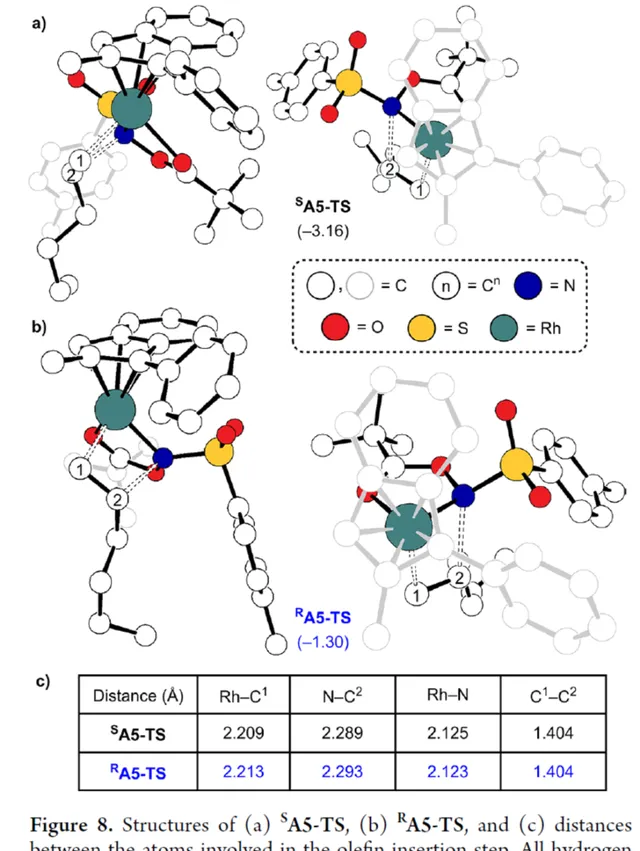
其次,配合物 A3 可與烯烴反應,生成配合物 A6 。由於 A3 的非對稱結構,它為烯烴提供了兩個不同的空位,這導致了對映選擇性的C−N鍵的形成( SA5 和 RA5 )。同時,( S )-產物的收率比( R )-產物高19-24倍。並且,發現這種遷移插入(第一個C−N鍵的形成)在整個催化迴圈中具有最高的活化能,它可能同時作為對映選擇性和速率決定步驟(Figure 7)。值得註意的是,反應的立體選擇性源於受質、磺醯胺和催化劑茚基配體上的苯基取代基之間的相互作用。同時, SA5-TS 和 RA5-TS 之間能量差的主要來自空間相互作用(Figure 8a)。
隨後,遷移插入後,中間體 A6 經氧化N−O鍵斷裂,生成中間體 A7 。利用 A7 中的缺電子環境作為驅動力, SA7-TS 和 RA7-TS 分別容易形成還原性的C−N鍵,生成中間體 A8 。最後,中間體 A8 透過解離後,生成目標產物,並再生活性催化劑 A1 ,從而完成催化迴圈的過程。
(圖片來源: J. Am. Chem. Soc. )
(圖片來源: J. Am. Chem. Soc. )
(圖片來源: J. Am. Chem. Soc. )
總結
南韓科學技術院Mu-Hyun Baik與美國埃默裏大學Simon B. Blakey課題組報道了一種平面手性銠茚基催化劑促進非活化末端烯烴的對映選擇性氮雜環丙烷化反應,並透過DFT計算揭示對映選擇性的起源。透過該策略合成了一系列對映體富集的手性氮雜環丙烷衍生物,並且在活化烯烴單元存在下對非活化的烯烴具有顯著的選擇性。計算研究表明,羥胺的活化發生在烯烴參與之前。氮賓和 π -烯丙基配合物的形成都不如醯胺中間體的形成有利。醯胺中間體經歷烯烴插入步驟,這既是對映選擇性決定步驟,也是速率決定步驟。對映選擇性源於烯烴和醯胺中間體之間的空間沖突的差異,這有利於從與烯烴結合的中間體形成( S )-氮雜環丙烷。
文獻詳情:
Patrick Gross, Hoyoung Im, David Laws, III, Bohyun Park, Mu-Hyun Baik,* Simon B. Blakey*. Enantioselective Aziridination of Unactivated Terminal Alkenes Using a Planar Chiral Rh(III) Indenyl Catalyst. J. Am. Chem. Soc. 2024 , https://doi.org/10.1021/jacs.3c10637











