必需基因特性表征與相關數據庫開發的研究進展
iMeta主頁:http://www.imeta.science
綜 述
● 原文連結DOI: https://doi.org/10.1002/imt2.157
● 2024年1月2日,天津大學高峰團隊在 iMeta 線上聯合發表了題為 「 Recent advances in characterization of essential genes and development of a database of essential genes 」 的研究文章。
● 本文總結了必需基因的相關研究進展,介紹了DEG數據庫及主要套用,並基於最新版本DEG 15.0進行了統計分析。應當註意的是必需基因是一個動態概念,而不是一個二元概念,這為必需基因未來的研究發展帶來了機遇和挑戰。
● 第一作者: 梁雅婷
● 通訊作者:高峰( [email protected] )
● 主要單位:天津大學理學院物理系、天津大學合成生物學前沿科學中心和系統生物工程教育部重點實驗室、天津化學化工協同創新中心
亮 點
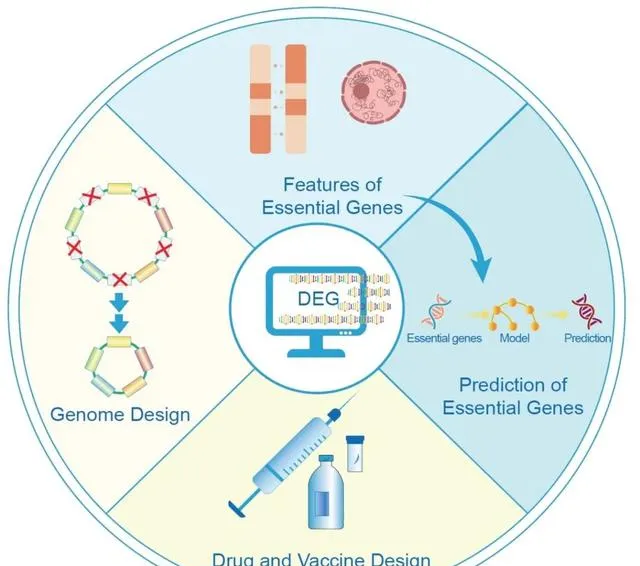
● 必需基因對生物體的生存和發育至關重要,目前已透過多種實驗手段鑒定出了大量生物體的必需基因,為研究它們的特性和開發預測演算法提供了寶貴的資源;
● DEG數據庫收集了大量必需基因的實驗結果,是開展必需基因相關研究的可靠資源,方便研究人員快速獲取特定物種的必需基因資訊,並在必需基因特征分析和預測、藥物和疫苗開發,以及人工基因組的設計和構建中進行套用;
● 必需基因的定義依賴於研究背景,而非簡單的二元分類。透過整合不同實驗結果,對必需基因進行定義和分類具有挑戰性,但也為探索基因相互作用機制等相關研究提供了可能性。
摘 要
在過去的幾十年裏,對必需基因的研究引起了人們廣泛的興趣。這些基因對於生物體在特定環境條件下的生存至為關鍵,在合成生物學和醫學領域具有重要的套用價值。隨著技術的不斷發展,已透過實驗手段獲得了越來越多關於必需基因的數據。與此同時,各種與必需基因相關的計算預測方法、數據庫和網絡伺服器相繼出現。為了促進必需基因的研究,我們實驗室建立了一個必需基因數據庫——DEG,為必需基因特征分析和預測、藥物和疫苗開發,以及人工基因組的設計和構建提供了重要參考。在本文中,我們總結了必需基因的相關研究進展,還介紹了DEG數據庫及主要套用,並基於最新版本DEG 15.0進行了統計分析。應當註意的是必需基因是一個動態概念,而不是一個二元概念,這為必需基因未來的研究發展帶來了機遇和挑戰。 Bilibili:
影片解讀:https://www.bilibili.com/video/BV1X5411i7Pt/
Youtube:https://youtu.be/j22ihy62Wb8
中文轉譯、PPT、中/英文影片解讀等擴充套件資料下載
請存取期刊官網:http://www.imeta.science/
全文解讀
引 言
必需基因是生物體在特定環境條件下生存所必需的基因,是分子生物學和遺傳學的重要概念之一。基因的必需性在相關的理論和套用研究中都十分關鍵。對必需基因的鑒定十分重要,因為這些資訊對生命科學、藥理學和合成生物學等領域具有實際套用價值。然而,基因的必需性受多種因素影響,包括生長條件、發育階段和遺傳背景等。在不同的實驗條件下,同一生物體可能展現出不同的基因必需性,且這些必需性可能隨著前進演化而發生變化。基因的必需性是一個動態的、需要持續評估的量化特征,特定條件下的必需基因的作用不應被忽視。
通常,必需基因的辨識依賴於實驗手段。1999年,透過在生殖道黴漿菌上進行全域轉座子突變實驗,首次確定了生物體在基因組水平上的一組必需基因。隨後,科研人員對各種生物體中必需基因展開了廣泛研究。辨識必需基因實驗技術的發展促進了必需基因數據的積累。辨識必需基因最常用的實驗方法包括單基因剔除、RNA幹擾、反義RNA、轉座子突變和CRISPR/Cas9等。除了實驗方法,必需基因的計算預測方法也相繼出現,進一步促進了必需基因的辨識。最常用的計算方法包括基於比較基因組學、基於約束的預測方法和基於機器學習的預測方法,它們為未來的必需基因研究提供了重要參考。
近年來,必需基因的研究已在各個領域進行。例如,了解必需基因的功能對於發現最小基因組的組成至關重要,這是合成生物學中重要的研究內容之一。研究必需基因可以加速構建具有特定表型特征的生物,並促進藥物和疫苗的開發。此外,它們在細菌生命中不可或缺的作用使病原體必需基因編碼的蛋白可以作為潛在的藥物靶標。因此,在多數細菌中保守的某些必需蛋白被視為有前景的廣譜藥物靶標,若僅在某種細菌中是必需蛋白,則為特定物種特異性靶標的可行候選。此外,還可以透過現有必需基因數據構建模型,來預測潛在基因的必需性。
隨著必需基因數據量的不斷增加,基於實驗或預測得到的必需基因數據的數據庫和線上服務應運而生,為必需基因相關研究提供了便捷可靠的參考。必需基因數據庫(DEG)於2004年建立,並隨著實驗數據的積累不斷更新。該數據庫囊括了從多種實驗方法獲得的全基因組規模的必需基因數據。到目前為止,DEG數據庫已成為必需基因相關研究的重要參考,根據Web of Science數據,該數據庫已累計被參照超過1100余次。
在本綜述中,我們簡要概述了必需基因相關的技術發展、數據庫等網絡服務的構建、必需基因的套用,並強調了基因必需性是特定於環境的概念。此外,我們還介紹了DEG數據庫在實際研究中的重要套用,並提出了未來DEG數據庫的發展方向。
必需基因的定義
必需基因對生物體的生存至關重要,因此被視為生命的基礎。然而,不同物種和研究中必需基因的估計比例存在顯著差異。同一物種內的不同研究顯示,某些基因在一個菌株中可能是必需的,而在另一個菌株中則不是;或者某些基因在一種生長條件下是必需的,而在其他條件下則不是,這表明基因的必需性不是其固有內容。造成這種變化的原因多種多樣,包括實驗方法、條件的不同,甚至實驗誤差的影響。因此,「必需基因」這一術語高度依賴於研究背景。只有當生物體存活的環境被精確定義時,一個基因才能被歸類是否為必需。細胞對特定基因或基因產物的依賴受其外部環境和遺傳背景的影響。因此,基因的必需性可能因這些因素而變化,並隨著每次刪除而變化。這些基因被稱為「條件性」的必需基因。例如,一些基因被辨識為「保護性必需」,因為它們在移除另一個基因後可能變得非必需。這通常是因為前者編碼了對後者基因毒性效應的保護功能。相反,第二個基因的喪失可能使一個非必需基因變得必需(合成致死)。合成致死最初是在研究果蠅和酵母時發現的,其中單獨失活兩個基因中的任何一個對細胞存活的影響都很小。然而,同時幹擾兩個基因或多個獨立基因的表達,包括突變、過表達和基因抑制,可以導致細胞死亡。此外,基因產物可以形成復合物,其中非必需基因有助於必需功能。例如,在涉及繁殖的出芽酵母的蛋白質編碼基因中,已辨識出五組潛在編碼具有必需功能蛋白質的非必需基因。營養條件也可以影響必需基因,因為攜帶失活非必需基因的突變菌株在最佳條件下可能對細胞表型產生最小或可忽略的影響,在次優條件下可能導致嚴重損害或生存能力喪失。然而,有證據表明在富營養培養基中生長的許多非必需基因對適應替代生長條件很重要。此外,不同的實驗方法可能產生不同的結果。例如,與基於RNA幹擾(RNAi)方法相比,基於CRISPR方法在人類細胞系中辨識出的必需基因更多。考慮到這些因素,基因的必需性可能是一個定量特征,而不是簡單的必需/非必需二分類,需要更加標準化的定量方法來衡量。研究表明,「條件性」的必需基因通常表現出類似於非必需基因的特征。這歸因於與其他必需基因相比,此類基因通常旁系同源基因數量較多,共表達水平較低,幾乎不編碼蛋白質復合物成分。這一結果解釋了「條件性」的必需基因和「永久性」必需基因之間的區別。基於這些特征,研究者已開發出一種隨機森林預測模型,用於辨識有條件的必需基因。
最近的研究發現,細菌和酵母可以透過基因組變化和適應力突變來適應環境,這些突變可以在條件性必需基因失活的情況下恢復細胞功能。這些研究表明,盡管失去了必需基因的功能,基因的必需性取決於細胞獲得突變和恢復增殖的能力。這表明必需性不再僅僅是基因內容,而是細胞內容,將必需性歸因於細胞途徑而非基因本身。這些發現為定義和研究必需基因提出了新的研究方向和挑戰。
必需基因的鑒定
目前,確定必需基因主要有兩種方法:實驗方法和計算方法。實驗方法可以在不同實驗條件下為必需基因提供具體的結果(圖1)。然而,這些方法可能成本高昂且勞動強度大。因此,利用電腦預測必需基因近年來得到了較快發展。
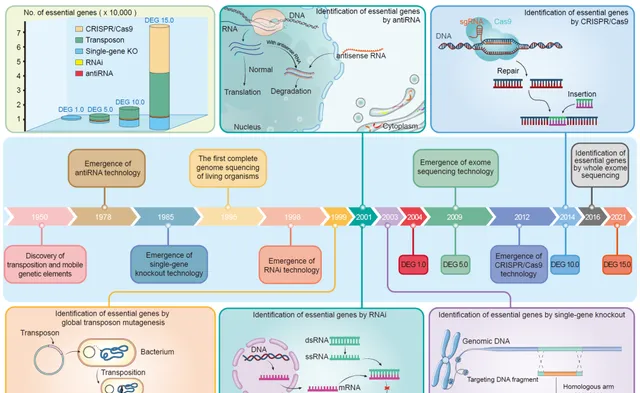
圖 1. 必需基因相關技術發展時間軸
此圖展示了一些重要的技術發展及其在必需基因研究中的最早套用。還顯示了DEG數據庫不同版本的更新時間,以及每個版本透過不同方法獲得的數據量。
基於實驗的方法
在分子生物學領域,鑒定必需基因有著悠久的歷史。1951年,Horowitz和Leupold提出蛋白質的一些主要成分可能對生命至關重要。在「基因組時代」之前,早期的突變誘導技術涉及使用化學或物理試劑在生物體的基因組中誘導隨機突變。透過誘導隨機化學突變並分析後代存活率來預測必需基因。例如,研究表明,大約50%、15%和12%的果蠅、秀麗線蟲和釀酒酵母的基因組是必需的。盡管如此,當時的實驗技術不足以鑒定具體的必需基因。在20世紀末,發展了轉座子技術、單基因剔除、反義RNA和RNA幹擾等技術,為基因層面的研究提供了更多可能性。1995年,首次獲得了流感嗜血桿菌和生殖道黴漿菌的完整基因組序列。獲得一個生物體的完整基因組序列是獲取完整基因列表的先決條件。在接下來的兩年中,大腸桿菌、枯草桿菌和釀酒酵母等模型生物的完整基因組也相繼公布。 1999年進行了第一個鑒定生物體必需基因集的實驗,該實驗透過全基因組轉座子突變驗證了生殖道黴漿菌生存所需的最小基因集。 轉座子插入檢測必需基因的原理基於轉座子的隨機插入及其對基因中斷的影響。透過分析轉座子中斷基因的插入位點和表達,可以確定必需基因及其對生長的影響。隨後,開發了透過針對單基因剔除、RNA幹擾和反義RNA來鑒定必需基因的方法,使研究人員能夠探索各種生物體中基因的必需性。單基因剔除實驗涉及移除一個基因以觀察表型變化。對於全基因組研究,這個過程必需重復多次,這需要全面的基因組註釋。RNA幹擾技術辨識必需基因的原理是,透過引入與目標基因信使RNA(mRNA)互補的小幹擾RNA(siRNA),形成雙鏈結構,進而引發目標mRNA的降解,從而抑制相應蛋白的表達。反義RNA技術辨識必需基因的原理是,透過引入與目標基因mRNA互補的單鏈反義RNA分子,這樣做可以與目標mRNA形成雙鏈結構,從而阻礙其表達,實作基因表現的抑制。
在必需基因研究的早期階段,主要關註點是微生物。動物基因幹擾的努力因動物組織培養中同源重組的固有低效而受阻。第一個突破是發現同源重組在來自小鼠囊胚的胚胎幹細胞中更為有效,促進了1989年第一只基因剔除小鼠的研究。2003年,研究者對秀麗線蟲進行了首次全基因組RNAi篩選,系統性地定義了突變體表型。隨後,這項技術迅速套用於哺乳動物細胞,幾個小組已經生成了涵蓋人類和小鼠基因組的RNAi庫,用於基因必需性篩選。RNAi已成為哺乳動物必需基因研究的主要方法。然而,不能忽略其非靶向效應和不完全的基因功能喪失限制。
下一代測序技術的出現使得快速獲取各種物種的全基因組序列成為可能。方便的測序數據存取促進了引入處理遺傳變異的其他方法。轉座子測序(Tn-seq)是一種新興技術,結合了轉座子突變和高通量測序,並使用在目標生物體中構建的高密度轉座子插入庫,透過高通量測序對其整個基因組進行功能分析。如TraDIS、INSeq、HITS和Tn-seq等技術已廣泛用於微生物中必需基因的檢測。Tn-seq的套用透過納入必需基因組元件(包括非編碼RNA),而不僅僅關註蛋白編碼基因,加深了對必需基因的理解。此外,Tn-seq可用於在體內、體外的各種實驗條件下篩選必需基因,而不局限於培養條件。Tn-seq顯著增加了在不同條件下鑒定的必需基因數量。
全基因組必需性篩選闡明了幾個生物過程的分子基礎。然而,基於RNAi的篩選常常受到非靶向效應和基因敲低而非完全喪失功能的困擾,這限制了我們對人類細胞中必需基因的了解。然而,CRISPR/Cas9系統的出現徹底革新了哺乳動物細胞基因組編輯。從根本上說,這個可編程的DNA內切酶由兩部份組成:源自鏈球菌的Cas9蛋白(或其他物種的類似蛋白),以及引導內切酶活性至目標DNA序列的單一導向RNA。CRISPR誘導序列特異性DNA雙鏈斷裂,導致移碼插入/缺失,從而導致蛋白功能的完全喪失。這項技術使得在酵母、植物和動物中進行經濟高效的基因編輯成為可能,對人類細胞編輯產生了重大影響。此外,Cas9的催化失活版本(dCas9)可以用於透過單一導向RNA定位特定DNA序列,這被稱為CRISPR幹擾(CRISPRi),或用於啟用基因表現,稱為CRISPR啟用(CRISPRa)。Cas9和dCas9已被用於繪制人類細胞系中必需成分的組成。2015年,三篇論文同時報道了 不同人類細胞類別中必需基因的全基因組鑒定 。此外,全外顯子測序是另一項突破,它使用序列捕獲或靶向擴增技術捕獲和富集整個基因組外顯子區域的DNA。與全基因組重測序相比,全外顯子測序主要針對外顯子區域的基因序列,具有更深入的覆蓋和更高的數據準確性,以快速鑒定體內的人類必需基因。
基於計算的方法
考慮到實驗方法的復雜性、高成本、勞動力和時間成本,計算方法通常被作為實驗方法的補充,以最小化必需性分析所需的資源。透過實驗鑒定的必需基因數量的增加為計算手段提供了參考。通常,有三種方法被用來確定基因的必需性:基於比較基因組學的、基於約束的和基於機器學習的方法。
最初,必需基因預測是透過依賴同源性的比較基因組學方法進行的。同源性對映發生在一個生物體內被復制的基因(旁系同源基因)或多個不同生物體中相關基因(直系同源基因)之間,這些基因是物種分化的產物。同源性對映透過比較多個生物體的序列,並根據一定的閾值確定它們的序列相似性。這種方法已被用於預測諸如黴漿菌、棒狀桿菌、瘧原蟲和布魯氏菌等物種的核心基因。一個主要挑戰是前進演化距離對比較基因組分析結果的顯著影響。盡管必需基因在細菌中通常具有高度的前進演化保守性,但跨物種的保守基因並不總是必需基因,這使得基於同源性的方法在預測必需性方面效果較差。以前的大規模分析顯示,只有少數基因在生命樹上是保守的,這意味著許多必需基因是物種特異性的。
基於約束的預測方法利用基因組規模的代謝網絡來闡明生物體內代謝途徑的生物學。這種方法依賴於基於基因組測序和註釋重建的代謝網絡的約束建模技術,以研究網絡的結構、功能和相互作用。通量平衡分析(FBA)是最廣泛使用的基於約束的方法,用於分析代謝網絡特性。它透過將質素平衡約束套用於化學計量模型,預測穩態條件下代謝物的流量。使用FBA預測必需基因涉及模擬基因剔除並評估其對網絡的影響。基於約束的模型已在三域系統的生物體上構建,並促進了基因必需性的研究。FBA的計算成本較低,因為它不需要動力學參數。然而,FBA也有顯著的局限性。首先,它只能預測代謝基因的重要性。此外,與能夠結合穩態和動態分析的能力不同,FBA需要酶動力學數據來評估瞬態條件下基因組規模代謝反應的活性。最後,FBA通常需要酶反應來解決代謝模型中的局限性,有時可能與實驗數據不一致。這取決於經驗建模,在某些情況下,參數預測可能具有挑戰性。
目前,最廣泛使用的必需基因預測方法基於機器學習演算法的構建,使用從必需基因的實驗結果分析中得出的特征。通常,必需基因的特征可以分為兩類:序列相關和背景相關(表1)。
表1. 必需基因的特征匯總
對必需基因特性的研究為使用機器學習預測必需基因提供了參考。通常,機器學習中預測模型的開發涉及以下步驟:構建訓練和測試數據集(必需/非必需基因數據)、特征選擇(必需基因的不同特征)、選擇和設計機器學習演算法,以及評估模型預測效能。各種研究已經使用基因組和蛋白質特征來開發和訓練用於預測基因必需性的分類器。多年來,已成功開發了基於基因組特征的多種演算法模型,用於理論鑒定必需基因。例如,範等人開發了一個結合必需基因蛋白質-蛋白質相互作用(PPI)和亞細胞定位的SCP演算法。這種方法結合了基於基因表現數據的改進PageRank演算法、加權亞細胞定位和加權亞細胞定位的皮爾遜相關系數。此外,還出現了預測非編碼區域必需基因的模型。張等人使用基於metapath-guided的隨機遊走開發了iEssLnc模型,這是第一個估算長非編碼RNA(lncRNA)基因必需性的模型。可以推斷出,準確預測需要「好」的數據和高效的機器學習技術。監督學習、半監督學習、無監督學習和強化學習是常用的機器學習技術。然而,基因必需性預測通常被建模為在監督學習下的分類問題。
深度學習是人工智能中機器學習的一個子集,其中神經網絡可以以無監督的方式從非結構化或未標記的數據中學習。最近,深度學習已被用於預測必需基因。例如,Deeply Essential是一個深度神經網絡,僅使用序列資訊來預測必需基因。與之前使用聚類和欠采樣數據集的方法相比,這個模型實作了更高的靈敏度和準確性。另一種必需性預測的深度學習模型采用了不同的方法,使用一個自動學習生物特征的框架,而不需要先驗資訊。這個網絡利用基因表現、亞細胞定位和PPI網絡的資訊來學習拓撲特征。然而,將深度學習套用於基因必需性預測有兩個主要缺點:(i) 深度神經網絡需要大量數據進行訓練,以優於傳統的機器學習演算法;(ii) 深度學習模型中調整超參數的過程復雜。
雖然套用機器學習方法方便,但它也面臨著一些挑戰,如預測質素難以衡量和無法在特定實驗背景下泛化。此外,考慮到必需基因的定義是特定於研究背景的,因此在定義訓練ML模型的標準時應謹慎,這取決於研究的目的。此外,特征的選擇和組合可能會影響預測效能,且沒有確定的方法來為不同的生物體選擇適當的特征。最後,對於研究不足的物種,選擇研究物種內數據受限於已知必需基因的數量較少,而使用跨物種數據可能導致準確性降低。
相關數據庫等網絡服務
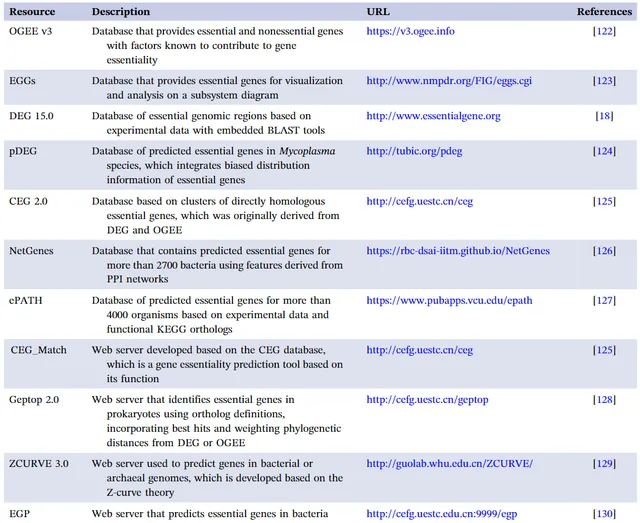
基於實驗數據和計算模型,建立了必需基因的線上數據庫等網絡服務(表2)。研究人員可以使用這些數據庫中的數據來研究必需基因/蛋白質的內在特性和與必需性密切相關的特征。除了基於實驗得出的必需基因數據庫外,還建立了儲存預測的必需基因數據的數據庫,以及一些開放存取程式,用於進行必需基因預測。
表2. 必需基因相關的數據庫和網上服務
必需基因的實際套用
在合成生物學領域的套用
必需基因負責維持細胞中正常的生理和代謝過程,使它們對構建具有高穩定性或特定功能的細胞至關重要。因此,這些基因為基因組設計等相關研究提供了理論基礎。目前,最小基因組是合成生物學中最重要的概念驗證之一。最小基因組是指維持生物體最必需生命活動所需的基因組;它一直是合成生物學領域的一個關鍵研究目標。建立一個維持生命所需的最小通用基因集可以極大地增強我們對生命最必需層面的理解,並在生產中具有實際套用價值。作為合成生物學的基石,必需基因可以作為構建最小基因組的參考。值得註意的是,必需基因和最小基因組的概念並不完全等同。必需基因代表了成功繁殖所需的一組基因,而最小基因組代表了維持細胞存活所必需的基因。在實踐中,鑒定和研究必需基因通常被用來推斷最小基因組的組成。然而,對細菌代謝網絡的計算建模表明,最小基因組所需的基因數量比所有必需基因的總和還要多。為了構建最小基因組,提出了自上而下的方法和自下而上的方法。自上而下的方法透過刪除隨機選定或未辨識的基因組片段來減小基因組大小。刪除可以透過各種實驗方法完成,如基於質體或基於線性DNA方法,以及利用特定位點的重組酶、轉座子和CRISPR/Cas9系統。透過逐步移除非必需基因和功能元件的自上而下方法,已經實作了幾個基因組的最小化。此外,研究人員還提出了一種名為MinGenome的自上而下基因組刪減演算法,該演算法從最長可能的刪減開始,連續刪除代謝和調控基因。為了避免致命或生長缺陷相關基因的刪除,該演算法透過對生物質產量施加限制來保留必需基因和合成致死對。自上而下方法的主要優點是從一個可操作的基因組開始,允許透過恢復到最後一個未刪減狀態來補救由刪除引起的任何不利影響。然而,這種方法耗時且可能導致意外的死胡同,因為在整個過程的每一步中,遺傳景觀都在改變,影響著其他基因的必需性。
自下而上的方法將基因片段與特定功能聯系起來。DNA合成、測序技術和基因組移植的進步使得合成具有復雜基因組的長DNA序列成為可能。該方法主要利用聚合酶鏈反應技術,使得重疊的短寡核苷酸池的組裝成為可能,並為自下而上方法提供了技術基礎。基於逐步減少基因組的幾輪合理設計和隨機突變,還有研究者構建了一個近似的最小細菌基因組JCVI-syn3.0,它比最初預測的個體必需基因集多出98個基因。這一觀察可以歸因於原始基因組中的非必需基因在基因組減少過程中的合成致死而變得必需或準必需。Breuer等人利用黴漿菌和細菌積累的代謝數據,並將其套用於通量平衡分析模型,建立了JCVI-syn3.0代謝網絡的計算模型,這可以更好地預測JCVI-syn3.0中的必需和準必需基因。在自下而上設計過程中,有必要闡明每個基因及其在整個遺傳背景中的相互作用的完整遺傳資訊。由於對基因組設計原理的了解有限以及目標生物體的復雜性,即使對於基因組較小的細菌,也存在眾多可能的基因組配置,這對該方法構成了挑戰。
正如上文所述,構建最小基因組的一個主要困難在於闡明整個遺傳背景中基因之間的相互作用,這對必需基因的鑒定和基因組構建的早期階段構成了重大挑戰。然而,電腦輔助方法可以透過表征具有各種類別基因組修改的細胞,加速產生與遺傳內容和細胞功能相對應的大規模數據,因此有可能拓寬和深化我們對整個細胞系統的理解,並有助於產生具有工業價值的生物系統。
在醫學領域的套用
傳統的藥物和疫苗發現方法資源密集且耗時。最近,減法基因組學和反向疫苗學被歸類為鑒定藥物和疫苗候選目標的強大方法。這些方法透過消除昂貴且耗時的試錯實驗的需求,簡化了藥物開發。鑒定潛在靶標是藥物和疫苗發現的第一步。考慮到必需基因的缺失或抑制可能對微生物產生致命效應,必需基因可以用作減法基因組學和反向疫苗學中藥物和疫苗目標的篩選標準。此外,在癌癥治療領域,合成致死性的概念已擴充套件到成對基因,其中一個基因由於缺失或突變而失活,另一個基因的藥物抑制導致癌細胞死亡,而正常細胞不受影響。在最直接的套用中,可以確定針對性治療,殺死缺乏特定腫瘤抑制基因的癌細胞,但保留正常細胞。此外,癌細胞系中必需但在人體組織中非必需的基因可以揭示與相應癌癥類別相關的致癌驅動因素、旁系基因表現模式和染色體結構。最近對大量癌細胞系進行的基因組規模CRISPR篩選的分析表明可能有數百個有效的藥物靶標,但絕大多數是特定環境的。此外,一些必需基因已與人類疾病相關聯。例如,透過基於人群分析的必需基因鑒定,人類大部份對功能喪失突變不耐受的基因屬於必需基因,並且已發現它們在人類減數分裂重組中發揮作用,可能促進某些疾病的發生。此外,基因必需性的資訊已被用於研究人類測序結果中與未知疾病相關的潛在致病基因變異。
然而,大多數現有的必需基因是在體外鑒定的,在體外和體內生長所需的基因之間可能存在顯著差異。一個創新的解決方案是在非傳統生長培養基中進行篩選,如在酸性培養皿中培育會造成肺部感染的銅綠假單胞菌。在篩選過程中優先考慮在營養有限培養基中表現活躍的產物對應的基因。在宿主模型中進行疾病篩選是另一種發現體內靶標的方法。例如,對被結核分枝桿菌感染的巨噬細胞進行高倍率顯微鏡篩選,鑒定了一系列針對細胞色素c的先導化合物。盡管對基因的系統研究揭示了眾多潛在靶標,但在靶標驗證過程中,通常需要對它們的功能有清晰的理解,這在鑒定藥物靶標時構成了挑戰。此外,藥物抗性的持續前進演化是不可避免的,需要進一步理解抗性機制。最後,必須承認基因必需性是一種可前進演化的特性,必需性程度最大的基因可能是有前途的藥物靶標。這在尋找藥物靶標時帶來了挑戰,但也帶來了機遇。因此,需要更多的研究來全面定義基因在不同環境和時間尺度下的必需性,並進一步探索必需基因的遺傳背景。
DEG數據庫的介紹
隨著必需基因數據量的不斷增長,迫切需要將這些數據組織成一個數據庫,以便於使用這些數據。因此,我們實驗室於2004年建立了DEG數據庫,並隨著實驗數據的積累不斷更新。該數據庫組譯了從多種實驗方法獲得的全基因組必需基因數據。最新版本的數據庫DEG 15.0,涵蓋了細菌、真核生物和古菌,於2021年釋出(https://tubic.org/deg)。總的來說,在DEG 15.0中儲存了由不同實驗方法確定的78組細菌、35組真核生物和2組古菌的必需基因數據集(圖2A和2B),並展示了每組必需基因的具體資訊(圖2C)。此外,DEG數據庫還包括與必需基因相關的分析模組,特別是針對細菌的,其中包含以下功能:(i)必需基因在先導鏈或後隨鏈上的分布;(ii)必需基因的亞細胞定位分布(圖2E);(iii)必需基因的同源群、EC號以及KEGG和GO富集資訊(圖2F,圖2G);(ⅳ)客製的BLAST搜尋工具,允許使用者進行特定物種和實驗的搜尋,以辨識已註釋或未註釋基因組中的必需基因(圖2D)。除了必需基因外,該數據庫還收集了一部份非必需基因的實驗結果,以及除了編碼蛋白質的必需遺傳元件之外的內容,如非編碼RNA和復制起始點。
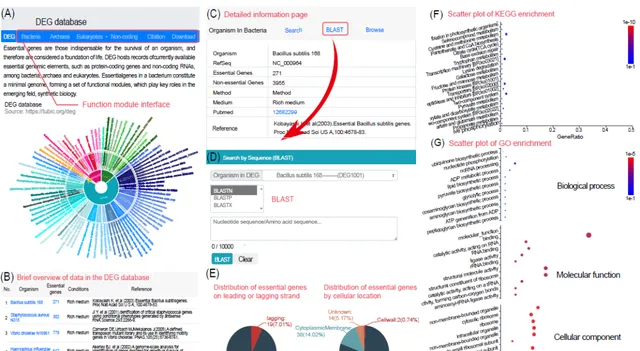
圖 2. DEG數據庫的概況
(A) DEG數據庫的主頁。提供了指向其他模組的介面,以及連結到不同物種中多個必需基因的實驗結果資訊。(B) DEG數據庫中所有數據的匯總頁面。(C) 特定菌株的實驗結果相關資訊截圖,包括菌株資訊、培養條件和參考文獻參照。(D) 相應菌株的BLAST搜尋界面。(E) 相應菌株中先導鏈/後隨鏈上必需基因的分布。(F) 相應菌株中必需基因的京都基因與基因組百科全書(KEGG)富集分析結果。(G) 相應菌株中必需基因的基因本體(GO)富集分析結果。
DEG數據庫的套用
DEG數據庫已成為與必需基因相關研究的重要參考資料,是Nucleic Acids Research數據庫專刊中最受歡迎的數據庫(Golden Database)之一。使用DEG數據庫可以快速存取特定物種的必需基因資訊。目前,DEG數據庫的套用主要集中在以下四個領域:人工基因組設計和構建、藥物和疫苗設計、必需基因特征分析,以及必需基因的預測(圖3)。

圖 3. DEG數據庫的主要套用
DEG數據庫的套用主要集中在以下四個領域:人工基因組設計與構建、藥物和疫苗設計、必需基因特性分析,以及必需基因的預測。
使用DEG數據庫中的必需基因數據進行基因組設計的研究通常是自上而下的方法,因為基因組的非必需區域通常被視為待刪除的候選區域。在自上而下的方法中,通常使用數據庫中的必需/非必需基因數據來辨識基因組中的刪除位置,以提高其穩定性。一般來說,包含非必需基因的區域是刪除的目標,而包含必需基因的區域通常被保留。此外,根據研究目的,還設計了刪除運輸蛋白基因、插入序列(ISs)、毒素-抗毒素對和其他功能元件。例如,劉等人選擇性地從伯克霍爾德氏菌株中刪除基因,以最佳化其基因組結構和生長速率。這項研究使用DEG數據庫中的非必需基因數據來促進高度保守的基因組縮減菌株的鑒定,增強了基因組工程策略的可預測性,並提高了菌株生產的效率和穩定性。實際上,DEG數據庫的數據也可為自下而上的基因組構建提供寶貴的參考。因此,DEG數據庫被推薦為研究細菌存活所需最小基因組的參考資源。
使用計算策略而非培養微生物來辨識潛在藥物靶標可以顯著降低時間和成本。DEG數據庫中的基因資訊已被用於鑒定病原細菌的必需蛋白。到目前為止,這種方法已被套用於確定包括假結核耶爾森菌、大腸桿菌、銅綠假單胞菌、索恩氏沙門菌、糞腸球菌、肺炎鏈球菌和假結核棒狀桿菌在內的病原體的潛在藥物靶標。此外,這一過程也適用於疫苗設計,稱為反向疫苗學。反向疫苗學可以顯著加速疫苗開發,因為它可以減少對單個抗原的廣泛經驗測試的需求。使用這種策略,已經鑒定了幽門螺桿菌、布魯氏菌病、傷寒沙門和金黃色葡萄球的潛在疫苗靶標。此外,疫苗設計全流程管道VacSol和PanR也整合了DEG數據庫中的數據用於輔助疫苗的設計。
DEG數據庫提供了必需基因特征分析的寶貴數據,從而有助於必需基因特征領域的研究。數據庫中包括非必需基因數據,使得可以進行兩類基因之間的比較研究。已有許多研究利用DEG數據庫提供的必需基因特征。例如,羅等人對細菌基因組進行了前進演化保守性分析,發現必需基因比非必需基因前進演化得更慢。必需基因在分布上的顯著性鏈偏差主要與功能性有關,與基本生物功能相關(如轉譯、轉錄和復制)的必需基因更偏向位於前導鏈上。令人驚訝的是,如果一個蛋白的特定功能域在多個生物中存在,則其必需性的可能性增加。此外,許多微生物的代謝網絡已根據必需基因重建,並在一定程度上使用自動化系統如SEED和BiGG模型進行管理。基於DEG數據庫和其他來源的數據,Magnúsdóttir等人系統分析了腸道微生物群的代謝相互作用以及外部因素對這些相互作用的影響。
在使用機器學習方法預測必需基因方面,DEG數據庫提供了一個理想的訓練數據集。例如,郭等人使用支持向量機(SVM)根據核苷酸序列數據衍生的Z曲線特征來預測人類基因。曾等人構建了一個基於深度學習的框架,利用蛋白質-蛋白質相互作用(PPI)網絡、基因表現數據和亞細胞定位資訊預測必需基因。DeepHE透過整合序列數據和PPI網絡的特征來預測人類的必需基因。施等人提出了一種名為iEsGene-CSMOTE的機器學習方法,該方法基於支持向量機,用於辨識必需基因,引入了一種基於聚類的過采樣技術CSMOTE,來克服數據不平衡的問題。除了利用必需基因特征外,周等人還提出了一種基於影像辨識預測必需基因的演算法,該演算法使用了一種帶有R-STDP學習規則的摺積脈沖神經網絡,透過辨識基於混沌遊戲表示得到的DEG數據庫中必需和非必需基因影像特征來預測必需基因。
基於DEG數據庫的分析
必需基因與非必需基因的比較
由於DEG數據庫15.0包含了大量已進行全基因組基因必需性篩選的細菌的必需基因和非必需基因數據集,因此它提供了探索基因組大小與必需基因數量之間相關性的可能。某些必需基因,如那些涉及復制、轉錄和轉譯的基因,編碼了所有基因組無論其大小如何都必需的必需細胞功能。因此,我們對DEG數據庫中包含必需和非必需基因的數據進行了統計分析。線性回歸結果顯示,非必需基因數量與基因組大小呈正相關,而必需基因數量相對恒定(圖4A)。無論基因組大小和實驗條件如何,沒有任何細菌物種的必需基因超過1000個。相對於基因總數,必需基因的百分比隨著基因總數的增加而降低(圖4B)。這一結果表明,必需基因的數量相對恒定,不隨基因組長度變化,而非必需基因的數量與基因組長度呈正相關。
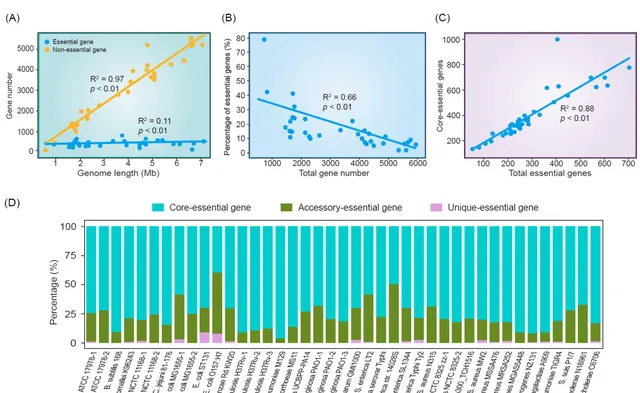
圖 4. 基於DEG數據庫中數據的統計分析
(A)基因組長度對應的必需基因和非必需基因數量。(B) 必需基因占總基因數量的百分比。(C) 核心必需基因數量與總必需基因數量之間的關系。(D) 全基因組框架下必需基因的分布。橫軸代表不同物種。特異、附屬和核心基因集中存在的必需基因分別用粉色、綠色和藍色突出顯示。縱軸代表不同類別必需基因的比例。
隨後,我們對DEG數據庫中必需和非必需基因的COG註釋結果進行了統計分析。在C類(能量生產和轉換)、F類(核苷酸轉運和代謝)、H類(輔酶轉運和代謝)、J類(轉譯、核糖體結構和生物合成)、K類(轉錄)和O類(轉譯後修飾、蛋白質轉換和伴侶蛋白)中,必需基因的數量多於非必需基因。然而,在G類(碳水化合物轉運和代謝)、N類(細胞運動)、M類(細胞壁、膜、被膜的生物合成)和U類(細胞內轉運、分泌和囊泡運輸)中,非必需基因的數量多於必需基。總之,COG功能富集分析表明,必需基因傾向於聚集在必需生命過程中,而非必需基因在環境適應和物質合成中扮演多樣化的角色和功能。
泛基因組分類中必需基因的分布
「泛基因組」這一概念最早Tettelin在2008年提出,指的是一個物種的所有基因的集合,可以分為「核心基因」(所有菌株共有)、「附屬基因」(兩個或更多菌株共有)和「特異基因」(特定菌株獨有)。構建一個物種的泛基因組能夠突破單一參考基因組限制,使得在物種層面上對基因組進行研究成為可能。泛基因組分類中的核心基因是物種內所有個體共有的基因,通常執行重要功能,編碼必需的生物學和表型相關性。考慮到必需基因和核心基因在生物體中的重要性,泛基因組分類已被納入當前必需基因研究中。例如, Saxena等人成功鑒定了使用SRB(硫酸鹽還原菌)模型的 Oleidesulfovibrio alaskensis G20的關鍵必需基因集,並對這些基因進行了泛基因組分類。結果表明,大多數必需基因屬於核心基因類別,而其他類別的必需基因可能是特定於環境的。此外,烏等人提出了一種策略,旨在減少枯草芽孢桿菌的基因組大小,同時確保保留核心和必需基因。
然而,目前缺乏大規模的泛基因組分類,特別是針對不同物種中必需基因的分類,以估計必需基因和核心基因的重疊程度。因此,我們提出了一種使用泛基因組框架對必需基因進行分類的方法。我們從NCBI下載了DEG數據庫中包含物種的所有完整細菌菌株。為了確保泛基因組分析的準確性,我們選擇了具有70個以上完整基因組的17個物種進行後續分析。首先,我們過濾N(未知)核苷酸超過1%的序列。接下來,我們排除了同一生物體的平均核苷酸同一性(ANI)值小於95%的菌株。ANI指的是兩個微生物基因組中同源片段的平均堿基相似度。同一物種內的生物體之間的ANI值通常≥95%。最終,我們獲得了與17個細菌物種相關的5900個菌株進行泛基因組分析。我們進行了泛基因組分析,並獲得了17個物種的泛基因組(包括核心、附屬和特異基因)的結果。透過將結果與DEG數據庫中相應菌株的必需基因數據使用BLAST進行比較,我們確定了泛基因組分類中必需基因的分布(圖4D)。
結果顯示,除了大腸桿菌O157:H7外,細菌中的大多數必需基因屬於核心基因。這表明,發揮重要作用的必需基因和核心基因在很大程度上重疊;這些類別的基因通常更加保守,被稱為核心必需基因。線性回歸分析顯示,核心必需基因的數量與必需基因總數之間存在顯著相關性(表4C)。然而,一些必需基因並不屬於核心基因,這可能與菌株對特定生長條件的適應有關。因此,這種分類將為必需基因的功能提供新的見解。此外,將泛基因組分析與必需基因研究相結合為疫苗和藥物的開發提供了新的視角。由於核心必需基因在更大程度上決定了病原體的生物學特征,辨識核心必需基因可能有助於設計有效的廣譜藥物。相比之下,物種特異的必需基因可能是特定菌株藥物的潛在靶標。
結論和未來展望
實驗技術的進步促進了大規模全基因組水平篩選必需基因的實作,為研究許多生物過程中涉及的必需基因提供了寶貴的見解,揭示了必需基因的復雜和多面性,為它們在合成生物學和醫學中的套用開辟了可能性。大量數據的產生也使得相關線上服務應運而生,例如相關數據庫和工具的出現,它們為必需基因研究提供重要參考與幫助。然而,正如本文所討論的,必需基因不是一個絕對概念,它取決於特定的環境和背景。由於分子相互作用的復雜性,使用計算方法,特別是機器學習方法來全面研究必需基因已被證明是具有發展前景的。在不久的將來,透過對具有不同理性設計基因組的細胞進行特征化,將加速產生有關遺傳內容和細胞功能相關性的數據,這最終將拓寬我們對整個細胞系統的理解與必需基因在各個領域的套用。此外,我們也將致力於不斷更新和維護DEG數據庫。除了收集有關必需基因的實驗數據外,我們還將關註必需基因的最新研究成果和新興發展趨勢。在未來,我們計劃在以下幾個領域增強和完善數據庫:首先,我們旨在更有效地評估必需性的程度。在數據庫的未來版本中,我們將添加透過實驗鑒定的條件性必需基因註釋來強調環境依賴性,將它們的必需性進一步基於在不同實驗條件下存在的可能性進行分級。其次,我們還將探索為相應的必需基因提供跨物種連結的可能性,以便於在不同菌株或物種直接進行必需基因的特征比較。此外,我們還將為必需基因添加泛基因組分析模組,該模組會在泛基因組框架內將必需基因劃分為核心、附屬和特異三個類別,以更好地在物種水平上研究特定必需基因。最後,我們還將加入更多與必需性相關的特征分析資訊,包括PPI(蛋白質-蛋白質相互作用)數據、表達譜和潛在藥物靶標等,並考慮添加更多視覺化手段以直觀地呈現獲得的結果。基於這一系列的更新,我們希望DEG數據庫能夠為必需基因研究提供更加全面、有價值的參考。
引文格式 :
Ya-Ting Liang, Hao Luo, Yan Lin, Feng Gao. 2024. Recent advances in characterization of essential genes and development of a database of essential genes. iMeta e157. https://doi.org/10.1002/imt2.157
作者簡介

梁雅婷(第一作者)
● 天津大學理學院物理系生物物理學專業碩士在讀。
● 研究方向為必需基因與泛基因組研究,相關學術成果發表於iMeta等期刊。

高峰(通訊作者)
● 天津大學理學院教授、博導,天津大學生物資訊中心主任。
● 主要從事微生物基因組生物資訊學與合成生物學研究。在Nucleic Acids Research、PNAS、iMeta等國際知名刊物上發表第一/通訊作者SCI論文68篇(Nucleic Acids Research、Genomics Proteomics & Bioinformatics、Briefings in Bioinformatics和Bioinformatics 系列25篇),其中52篇為唯一第一/通訊作者,獲Science、Nature等論文參照並佐證,相關成果得到中央電視台、【科技日報】、【人民日報】等國家級媒體報道。先後主持國家自然科學基金專案(5項)、國家重點研發計劃課題(課題經費1091萬)。












