水平有限,公众号翻译的文章可能不能完全代表作者的观点。

摘要
多倍体是真核进化中的重要机制,在植物界尤其普遍。然而,我们对这种现象及其对进化的影响的了解仍然有限。 多倍体研究的一个主要障碍是很难解开异源多倍体的起源。 由于杂交和复杂的后多倍体二倍体化过程的综合作用造成基因组的剧烈变化和异源多倍体信号的侵蚀, 解决异源多倍体的起源长期以来一直是一项具有挑战性的任务。 在这里,我们以款冬亚族(菊科:千里光亚科)这个有趣的案例重新审视这个问题,并通过开发HomeoSorter,通过将同源基因与亲本亚基因组进行定相分析,建立了一条新的网络推理管道。该管道基于先前研究的基本思路,但进行了重大更改以便解决扩展问题并实现一些新功能。通过模拟数据,我们证明 HomeoSorter在基因组规模的数据上能高效运行,并且在识别多倍体模式和分配同源基因方面具有很高的准确性。 利用Phylonet的最大伪似然模型HomeoSorter和基因组规模数据,我们进一步解决了款冬亚族的复杂起源问题,这是一个物种类群(约45属和710种),其特征是具有高碱基染色体数(主要是x=30、40)。特别是,推断的模式得到了染色体证据的有力支持。款冬亚族被发现包含两个具有连续异源多倍体起源的大类群:款冬亚族(主要是x=30)和类款冬亚族组(x=40)。两个异源多倍体事件首先产生了款冬亚族,第一个事件发生在千里光亚族的祖先(x=10)和与短舌菊属的类群相关的谱系(很可能x=10)之间,以及由此产生的杂交谱系与山绒菊属(x=10)的祖先杂交,并导致款冬亚族的产生。然后,在早期多样化之后,款冬亚族的中美洲群体(主要是x=30)再次与山绒菊属谱系一起参与第三次异源多倍体事件,并产生了类款冬亚族群体。我们的研究强调了 同源排序器和同源排序方法在多倍体系统发育学中的价值。款冬亚族具有丰富的物种多样性和清晰的进化模式。和类款冬亚族群也是未来多倍体研究的优秀模型。
关键词: 异源多倍体;短舌菊属;绒安菊属;同源排序;网状;目标富集;款冬亚族
引言
多倍体是植物进化中重要且普遍的机制 (VandePeer等人2017;Clark&Donoghue2018;Fox等人2020)。它 被普遍认为是遗传新颖性的来源(Soltis和Soltis2016)以及许多进化变化的驱动因素 ,包括生态位转移(teBeest等人,2012;Smith等人,2018)、物种多样性的增加(Edger等人).2015;Tank等人,2015),以及灾难情况下的生存(Fawcett等人,2009;Vanneste等人,2014)。目前,在植物进化史上已鉴定出超过180个古代多倍体事件(Ren等人,2018年;一千个植物转录组计划2019年),并且估计24-35%的现存维管束物种是新多倍体(Wood等人,2019年;Mayrose等人2011;Barker等人,2016)。通常,多倍体分为同源多倍体和异源多倍体,基因组复制分别发生在同一物种内或通过不同物种之间的杂交产生(Stebbins1947;Soltis等人2014b)。虽然同源多倍体的发生率先前被低估了(Ramsey和Schemske1998;Soltis等人2007;Parisod等人2010;Barker等人2016),但 异源多倍体被广泛认为是主要类型,并在进化中发挥更重要的作用 (例如,Soltis等人2003;CoyneandOrr2004;Mallet2007)。
尽管异源多倍体很普遍且很重要,但解开异源多倍体的起源长期以来一直是一项具有挑战性的任务(Oxelman等人2017;Rothfels2021)。这首先是由于异源多倍体基因的复杂进化。多倍体之后通常是二倍体化过程,其中涉及复杂的基因组变化,例如基因丢失和染色体重排,随着时间的推移逐渐侵蚀多倍体信号(Wolfe2001;Soltis等人2015;Li等人2021)。异源多倍体结合了多个不同的基因组,通常会导致更剧烈的基因组变化,称为「基因组休克」(McClintock1984;Bird等人2018;NietoFeliner等人2020)。此外,其他进化过程,包括基因重组、水平基因转移和不完全谱系排序(ILS),可能会使异源多倍体的网状模式进一步复杂化(Steenwyk等人2023)。其中,ILS是系统发育不一致的一个特别常见的原因,并且由于它们相似的不一致模式而很难与杂交区分开来(Degnan和Rosenberg2009;Degnan2018)。因此, 异源多倍体基因往往经历非常不同的进化历史,使得很难恢复异源多倍体的真实进化模式。
普遍缺乏高效可靠的网络推理工具也极大地阻碍了异源多倍体的研究。传统上,异源多倍体是根据叶绿体和核基因树之间的细胞核不一致推断的。测序技术的快速进步彻底改变了这一点,测序技术使我们能够对基因组中大量未连锁的基因座进行采样,并恢复尽可能多的基因拷贝。这两个方面对于识别可靠的异源多倍体模式都至关重要(Oxelman等人,2017)。然而,大数据也对系统发育分析提出了重大挑战,需要分析工具不仅计算效率高,而且能够解释复杂的进化过程。目前,该领域最广泛使用的程序是Phylonet中实现的最大伪似然(MPL)模型(Yu和Nakhlep015)(Than等人,2008年;Wen等人,2018年)。它可以复杂地解释杂交和ILS,并且通常具有很高的计算效率(Yu和Nakhlep015)。另一个近年来流行的程序是Phylonetworks(Solís-Lemus等人2017),它也是基于MPL模型,具有与Phylonet类似的性能。然而,其他程序都有很大的局限性。例如,HyDe(Blischak等人2018)可以测试特定的杂交假设,但无法估计明确的进化历史。PhyDS(Edger等人2019)只需计算异源多倍体等位基因的姐妹即可推断亲本谱系。PhyLiNC(Allen-Savietta2020)和NetRax(Lutteropp等人2022)牺牲了ILS以实现更快的计算速度。这些计划无疑过度简化了潜在的进化过程,而且它们的结果通常不太令人信服。
最近,异源多倍体系统发育学的一个新兴方向是对整个基因组中的等位基因进行定相,即将同源物分配给其亲本亚基因组(Oxelman等人2017;Rothfels2021)。除了解决异源多倍体的亲本问题外,该策略最有趣的特征是它可以产生分叉的多标记树(而不是网络),在该树上异源多倍体由多个亚基因组代表,这些亚基因组与各自的潜在祖先细胞聚集。通过分阶段同源物和分叉树,我们可以进一步进行一些以前对于异源多倍体来说困难或不可能的下游分析(例如,网状事件的年代测定),只需使用现有的成熟工具(例如,BEAST)。
提出的同源排序方法(Bertrand等人,2015年;Oberprieler等人,2017年;Lautenschlager等人,2020年;Šlenker等人,2021年;Nauheimer等人,2021年;Freyman等人,2023年;Leal等人,2023年)),Oberprieler等人的观点。(2017)看起来简单而有研究前景。该方法使用排列策略和物种树重建方法将同源物分配给亲本亚基因组,并最终生成整体多标记物种树(Oberprieler等人2017)。该方法由三个步骤组成。首先,它为每个基因和多倍体生成所有可能的等位基因组合,并使用Phylonet中最小化深度合并的方法(MDC;Yu等人2013)对每个组合进行树重建。最小化深度合并的组合被认为最适合数据并被选择。然后,基于这些选择,该方法为每个多倍体的所有基因生成所有可能的亚基因组组合,并执行相同的树重建以选择最佳组合。最后,将所有多倍体样本的选择组合起来,产生总体最优的多标记物种树。Oberprieler等人使用这种方法。(2017)成功解决了具有复杂时间场景和不同类型多倍体的模拟和经验案例。然而,由于在第二步中对所有基因的所有可能的亚基因组组合进行了详尽的搜索,该方法只能适用于小型数据集。对于大型数据集,计算非常昂贵甚至不可能,因为子基因组组合会随着基因数量的增加而呈指数增长。Lautenschlager等人(2020)试图通过开发AllCoPol来解决这个问题,采用启发式算法来提高计算效率。然而,问题仍然存在,因为他们仍然试图同时计算所有基因座。预计进一步的发展会将这种有方法被应用于大型数据集。
在这项研究中,我们通过来自菊科千里光族的款冬亚族的有趣案例来探讨异源多倍体系统发育问题。千里光科是菊科中最大的族群之一,约有150个属和3500个物种,分布几乎遍及世界各地(Nordenstam,2007;Nordenstam等人2009)。该部落的一个显著特点是多倍体发生率极高,约80%的物种是多倍体(Ren等,2021)。款冬亚族作为第二大亚族,是千里光亚科中最突出的多倍体类群。它包括约45属和710种,主要分布在东亚和美洲(Nordenstam2007;Nordenstam等人2009)。除了3个小属(黏子菊属、腋绒菊属和细叶垫菊属)有8个物种的基本染色体数x=9(很少为7)外,所有物种均基于x=30或其衍生物(Nordenstam)1977年;诺登斯坦等人,2009年。值得注意的是,五个属(赤道菊属、绒安菊属、南赤道菊属、藤蟹甲属和拟绒安菊属约164个南美洲特有种(Nordenstam,2007),统称为类款冬亚族类群(Jeffrey1992),具有更高的基础染色体数x=40(Robinson等人1997)。
在之前的一项研究中,我们开发了1104个单/低拷贝核(SLCN)基因用于千里光亚科的系统发育学研究(Ren等人2021),并纳入了一些款冬亚族物种进行测试。与二倍体千里光亚科物种通常每个基因仅恢复一个序列相反,款冬亚族样品通常具有两个或三个旁系同源拷贝。此外,对于大约一半的基因,款冬亚族序列形成两个或三个不同的、强有力支持的进化枝,每个样本的旁系同源物通常均匀地分成不同的进化枝。在这些分支中,一个似乎总是与山绒菊属聚集在一起,另外一两个与千里光族、千里光亚族、短舌菊属的类群和头状千里光属的美德利 - 伍德氏头状千里光(Caputia medley-woodii (Hutch.) B.Nord.))有不同的关系。这些结果强烈表明款冬亚族的异源多倍体起源。然而,该研究(Ren等人2021)仅旨在选择SLCN基因,并没有包括足够的款冬亚族代表。此外,与短舌菊属的类群(另一个基于x=30的潜在异源多倍体群体)的关系非常复杂,需要更仔细的检查。因此,我们没有在该研究中进一步讨论该问题。
这项研究有两个主要目的。首先是为异源多倍体系统发育学 提供一种新的高效同源排序程序 。这是通过重塑Oberprieler等人的方法来完成的。(2017)解决扩展问题并实现一些新功能。第二个目标是自信地 解决款冬亚族的异源多倍体模式。 为了实现这一目标,我们通过合并许多新样本来扩展我们之前的工作(Ren等人2021),以更好地涵盖千里光亚科的多样性。使用基因组规模数据(1104个SLCN基因;Ren等人,2021)、我们的新程序和Phylonet的MPL模型,我们彻底研究了款冬亚族的起源。
材料与方法
分类单元采样
我们首先采用了之前研究中的26个物种的序列数据(Ren等人2021)。这些包括千里光族主要分支的24个物种,多榔菊族的狭舌多榔菊(Doronicum stenoglossum Maxim.),金盏花族的金盏花(Calendula officinalis L.)。千里光族的串珠千里光(Curio rowleyanus (H.Jacobsen) P.V.Heath)和向日葵族的向日葵(Helianthus annuus L.),它们是我们之前基因选择的基础(Ren等人,2021)。在这里,我们另外采样了29个物种,以更好地涵盖千里光亚科的多样性。特别关注二倍体属(即具有2n≤20任何可靠染色体报告的属),这些属的采样相对密集,以便更准确地确定款冬亚族的亲缘关系。这29个物种包括代表一个重要基部分支(在艾伯塔菊属(垫菊属 Cass.)之后第二个分化)的板叶卡佩利奥菊(Capelio tabularis (Thunb.) B.Nord.),该分支在我们之前的研究中未曾涉及;有茎头状千里光(Caputia scaposa (DC.) B.Nord.),这是一个孤立属分支中的第二个物种;来自千里光亚族15个另外属的15个物种;来自短舌菊属类群另外3个属的3个物种;代表类款冬属组的裸叶金鸡菊(Gynoxys psilophylla Klatt);以及来自款冬亚族另外8个属的8个样本。除此之外,还从菊科的其他主要谱系中取样了19个其他物种,以进一步测试和过滤家庭环境中选定的SCLN基因。本研究总共包含76个样本,详细信息参见Dryad上的补充表S1(https://doi.org/10.5061/dryad.573n5tbf7)。
实验方案
Ren等人选择的1104个SLCN基因。(2021)用于本研究。Ren等人采用的26个样本。(2021)已经有了序列。串珠千里光和向日葵的序列分别检索自OneKP(www.onekp.com;代码:BMSE)和GenBank(登录号:GCA_002127325.1;Badouin等,2017)。在这里,我们生成了48个新样本的数据。靶标富集的实验室方案严格遵循Ren等人的实验方案。(2021)。使用改良的CTAB方案(Doyle和Doyle1987)从硅胶干燥的叶子或植物标本中提取基因组DNA,并在QsonicaQ800R2(美国康涅狄格州纽敦)中超声处理至300-400bp。使用NEBNext试剂盒(Ultra II DNA 文库制备试剂盒E7645和多重寡核苷酸E6440;新英格兰生物实验室,IP交换机,马萨诸塞州,美国)制备DNA文库,然后使用Ren等人使用的相同定制探针进行溶液内序列捕获。(2021),其是基于单独的外显子设计的,参数为120聚体诱饵和6X平铺密度。按照制造商的方案进行八个捕获反应,每个反应从六个DNA文库中均匀混合总量500ngDNA,杂交在65°C下保持40小时,最后进行14个PCR扩增循环。使用Qubit3.0荧光计(赛默飞世尔科技,Waltham,马萨诸塞州,美国)和配备高灵敏度D1000ScreenTapes的Agilent2200TapeStation系统(安捷伦科技公司,Santa Clara,加利福尼亚州,美国)评估DNA浓度并对文库进行定量。在Novogene(美国加利福尼亚州萨克拉门托)的Illumina HiSeq 4000平台(美国加利福尼亚州圣地亚哥)上使用双端150bp进行测序。
基因组装
原始读取首先在Trimmomatic 0.38版本中进行清理(Bolger等人2014),通过删除最多两个不匹配的接头、30的回文剪辑阈值、10的简单剪辑阈值、检测甚至单个接头碱基,并保留短片段的两个读数。前导5个和尾随15个低质量碱基也被去除,并以4个碱基窗口大小和15的平均质量阈值进行滑动窗口修剪。短于40的清理读数被进一步丢弃。生成的配对读数在FastQC 0.11.9版本中合格(可在https://www.bioinformatics.babraham.ac.uk/projects/fastqc/获取)并用于基因组装。
1104个目标基因在HybPiper 1.3.1版本中组装(Johnson等人2016)通常使用默认设置。它包括使用BWA 0.7.1版本(Li和Durbin2009)将读数分配给目标基因,SPAdes 3.12.0版本(Bankevich等人2012)用于组装重叠群,以及Exonerate 2.2.0版本(Slater和Birney2005)用于将重叠群与参考比对并确定外显子-内含子边界。
SLCN基因的旁系同源性和过滤
就像我们之前的研究(Ren等人2021)一样,许多基因和样本在基因组装过程中都发出了旁系同源警告。继Ren等人之后(2021),我们重新评估了家族背景下的所有基因,并排除了那些在不同部落中表现出同源性的基因。检索了HybPiper识别的所有潜在旁系同源物。对于没有旁系同源警告的样本,我们使用最长的从头组装的重叠群,而不与其他重叠群连接,从而降低了嵌合体的风险。这些序列与MAFFT 7.407版本中的串珠千里光和向日葵的序列进行比对(Katoh和Standley2013)采用L-INS-I精度导向方法,然后在trimAl 1.4版本中进行修剪 (CapellaGutiérrez等人2009)具有自动严格模式。然后进一步排除剩余核苷酸少于300个的序列(不计算缺口)。各个数据集已提交至RAxML 8.2.11版本(Stamatakis2014)用于最大似然(ML)分析,全部采用GTR+GAMMA模型和20次搜索重复。使用ETE3工具包评估基因树(Huerta-Cepas等人,2016)。仅保留那些所有部落在单个基因树上都是单系的基因。
此外,为了尽量减少缺失数据的潜在影响,进一步排除了缺失程度较高的基因。具体来说,我们通过保留超过41个(>75%)千里光科样本(包括串珠千里光)、19个(>75%)款冬亚族和短舌菊属的类群样本以及11个(>50%)非千里光科样本来过滤基因(包括向日葵)。这里可能值得注意的是我们只采用Ren等人的26个物种的原因。(2021)用于这项研究。Ren等人使用的48个物种中(2021),27属于Ligularia-Cremanthodium-Parasenecio(LCP)复合体(Liu等人2016),它是款冬亚族的一个主要分支,主要分布在东亚(Pelser等人2007;Ren等人2021)。该复合体被用作示例来测试所选SLCN基因在通用和特定水平上的系统发育效用,并且采样相对密集。如果包含所有27个样本,则该基因过滤中的偏差将是不可避免的,与其他款冬亚族群体相比,LCP复合体显著偏向于LCP复合体。因此,我们只使用了五个样本来代表这个复合体,其中五个最大的属各一个。这一数字与款冬亚族其他主要类群的抽样数量相似(3-5个,但雌性类群只有一个可用)。最后,我们的过滤结果留下了727个基因。这些是以下所有分析的基础,以下称为G727数据集。
千里光族主要谱系的鉴定
由于网络推理的计算量很大,因此我们必须进行二次采样才能进行分析。首先生成了一个初步的系统发育框架,以验证先前确定的千里光亚科的主要谱系(Pelser等人2007,2010)。这里使用了G727数据集,但丢弃了所有潜在的旁系同源(即,仅使用了HybPiper建议的「主」副本;以下称为G727_Hyb数据集)。超矩阵和多物种合并方法都被用来推断系统发育。对于超级矩阵方法,使用与HybPiper一起分发的fasta_merge.py脚本连接基因,然后在RAxML中进行分析。各个基因被分配为单独的分区,全部采用GTR+CAT模型。使用具有100个引导复制的快速引导算法来评估节点支持。对于多物种合并方法,首先使用RAxML与GTR+GAMMA模型和100次快速引导复制生成单个基因树。然后,跟随Zhang等人(2018)和Ren等人(2021),Newick Utilities中支持度低于30%的分支被折叠(Junier和Zdobnov2010),以提高物种树推断的准确性。使用ASTRAL-III5.7.3版本中总结了物种树(Zhang等人2018),节点支持通过局部后验概率来测量(SayyariandMirarab2016)。
系统网络分析
首先使用Phylonet 3.8.2版本中的MPL模型(Yu和Nakhlep015)研究了款冬亚族的网状进化(Than等人,2008年;Wen等人,2018年)。该模型可以推断显式网络,同时考虑ILS,并且可以处理相对较大的数据集(Yu和Nakhlep015)。然而,考虑到千里光科中多倍体的普遍存在,本研究中约70%的千里光科样品是多倍体,并且可能具有独立的起源。在一次分析中不可能分析所有这些。因此,我们采取了一步一步的方法,每一步都进行不同的子采样,以最终阐明款冬亚族的起源。
这里使用了G727数据集的各个基因树,全部使用Newick Utilities进行植根。此外,支持度低于30%的分支被折叠,这一策略被证明可以改善ASTRAL分析中的树推理(Zhang等人2018;Ren等人2021),并且在这里也进行了尝试。
第一个分析(以下称为Phylonet_A1)是为了了解千里光亚科的基本网状图案。选择了22个样本来代表上述千里光科的主要谱系,包括代表四个不同二倍体谱系的四个物种(艾伯塔菊属的凹缘艾伯塔菊(垫菊属 emarginata (Gaudich.) Cass.)、卡佩利奥菊属的板叶卡佩利奥菊(Capelio tabularis)、切尔索多马属的白色切尔索多马(Chersodoma candida Phil.)和头状千里光属的美德利-伍德氏头状千里光(Caputia medley-woodii (Hutch.) B.Nord.))、短舌菊属联盟的莫罗短舌菊(短舌菊属 monroi (Hook.f.) B.Nord.)、代表款冬亚族六个主要组的六个物种,以及千里光亚族的11个物种(从二倍体到十倍体)。ETE3用于修剪单个基因树。由于很难确定真实的网状结构数量(Cao等人2019),因此进行了六次测试,分别允许1到6个网状结构事件。任何测试均未指定杂交物种(即未使用「-h」标志)。
由于Phylonet_A1的结果表明款冬亚族谱系出现在现存千里光亚科的多样化之前,因此千里光亚科多倍体可能与款冬亚族的起源没有直接关系。为了进一步检验这一假设,进行了14项额外分析(Phylonet_A2–A15),每项分析都研究了千里光亚科的多倍体样本。类群采样在Phylonet_A1的基础上进行了调整,非千里光亚科样本不变,千里光亚科采样替换为其全部二倍体样本加1个多倍体样本。对于每次分析,允许网状结点的数量最多为所研究物种的倍性水平的一半(即比潜在网状化事件多一个)。例如,对于番千里木属,2n=10x=100,通常预计会出现最多四个网状事件,我们进行了五次测试,允许最多五个网状结构以获得更可靠的结果。此外,与Phylonet_A1不同的是,所研究的千里光亚科多倍体需要通过「-h」标志指定为杂种;否则,程序将始终首先推断款冬亚族的网状结构。
在确认千里光亚科多倍体与款冬亚族的起源无关后,我们排除了所有千里光亚科多倍体,并更彻底地研究了款冬亚族的网状模式(Phylonet_A16)。对于分类单元采样,我们保留了四个二倍体物种来代表千里光亚科,并将款冬亚族和短舌菊属的采样扩展到每个主要组的三个样本,但雌性类组中只有一个可用。进行了六次测试,分别允许一到六次网状事件,并且所有测试都没有指定杂交物种。
在所有上述分析中,六倍体短舌菊属的类群尚未推断出任何杂交信号,并且款冬亚族五个主要类群的亲本谱系通常与该类群直接相关。为了证实这种模式,我们针对短舌菊属的类群(Phylonet_A17)进行了额外的分析。分类单元采样是根据Phylonet_A16进行调整的。短舌菊属的类群的四个样本都包括在内。绵毛千里光,千里光属的第二个物种,与短舌菊属的类群关系最密切。款冬亚族的样本被简化为五个物种,除了不相关的雌性类群外,每个主要类群各有一个。类似地,进行了六次测试,分别允许一到六次网状事件,并且所有测试均以短舌属联盟和节肢亚科设置为杂交种。
为了获得可靠的结果,所有上述测试重复十次,每次搜索重复20次(「-x20」)。具有最高对数伪似然值的结果被认为最适合数据。
同源物排序
为了促进同源分选方法在基因组时代的使用,并为款冬亚族的网状进化提供额外的证据,我们基于Oberprieler等人的基本思想开发了一种新的流程(2017)但有重大变化。如上所述,Oberprieler等人(2017)试图一次计算所有基因的所有子基因组组合,这对于大型数据集来说是不切实际的,因为子基因组组合会随着基因的增加而呈指数增加。在这里,我们引入了一种分层方法,其中我们逐渐将基因分组在一起,并在每一步中为每个组选择局部最佳组合,直到所有基因最终组合以确定总体最佳亚基因组组合。
除了这个改变之外,我们还做了一些其他的重大调整。首先,我们 使用ASTRAL-III 5.7.3版本(Zhang等人,2018)和ASTRAL-Pro 1.1.2版本(Zhang等人2020)代替用于树推理的MDC模型 。与简约的MDC模型相比,ASTRAL程序在解释ILS方面更加复杂(Zhang等人2018,2020)。尽管Phylonet也有复杂的模型(例如最大似然和MPL),但ASTRAL程序仍然是首选,因为它们的计算速度更快。其次,我们放弃了Oberprieler等人(2017)方法的第一步,即单独分析基因,并且对于每个基因和样本,每个亚基因组最多只允许有两个等位基因。相反,我们 将基因树分成小组,并将这些小组视为最基本的分析单位。 此外,将等位基因分配给亚基因组没有限制(即,一个或所有等位基因可以分配给同一亚基因组);这可能有助于减轻进化噪音或PCR扩增和/或测序过程中引入的一些错误的潜在影响。第三,在分析多个多倍体时,Oberprieler等人。(2017)分别研究了它们,仅在最后一步合并结果。然而,我们试图尽 可能全面地分析它们(见下文),因为它们的进化历史可能密切相关,应该作为一个整体来考虑。
下面我们更详细地描述了管道。首先,对输入的个体基因树进行格式化,主要是为多倍体种质附加独特的基因索引,防止在生成等位基因/亚基因组组合过程中发生混合。然后,通过排除所有其他多倍体,对每个研究的多倍体的输入树进行修剪。第三步包含我们管道的核心过程,图1中通过一个简单的示例进行了说明。它由多个子步骤组成,每个子步骤都涉及两轮ASTRAL分析。在第一个子步骤中,根据限制每组中每个多倍体的等位基因总数的标准(即「--allele」标志),将各个基因树分成小组。例如,图1中的前三棵树都具有一个多倍体(红色)的两个等位基因和第二个多倍体(蓝色)的两个等位基因。如果「--allele」设置为「4」,则只有前两棵树可以分组在一起,因为如果也包括第三棵树,第一个多倍体的等位基因总数将超过限制。对基因进行分组后,我们为每组中的每个多倍体生成所有可能的等位基因组合,并使用ASTRAL-III为它们重建树。选择最终归一化四重奏得分最高的前几个(「--最佳」)组合(如果多个组合得分相等,则随机选择一个或多个组合)。然后在第二轮ASTRAL分析中对它们进行重新评估,并考虑所有多倍体。所有可能的亚基因组组合都是通过第一次ASTRAL分析中选择的所有多倍体组合的排列生成的,然后使用ASTRAL-III进行计算。选择对整体最优做出贡献的一个。
在第二个子步骤中,每几个基因组(「--every」)被汇集在一起作为一个更大的组。对于每个较大组中的每个多倍体,所有可能的亚基因组组合都是根据前一个子步骤的结果生成的,并在ASTRAL-Pro中分析前几个(「--最佳」)组合。然后在ASTRAL-Pro中重新评估选定的组合,以确定有助于整体优化的组合。图1所示的示例很简单,整个过程只需要两个子步骤。对于大型数据集,第二个子步骤可能需要重复多次才能最终将所有基因分组在一起以生成最终的最佳多标记物种树。
应该注意的是,我们在第一个子步骤中使用了ASTRAL-III,在其余子步骤中使用了ASTRAL-Pro。这是因为这两个程序虽然基于相似的算法,但执行起来却截然不同。根据我们的测试,ASTRAL-III在辨别竞争性多标记树之间的差异方面更加敏感,这在最初的等位基因到亚基因组分配中尤为重要,但其计算效率随着等位基因的增加而大大降低。ASTRAL-Pro可以更有效地处理大型数据集,因此被用作替代品。
由于我们的新管道的一些缺点和随机问题(参见讨论),不同运行的结果可能会有所不同。可以运行多次重复(「-r」)来总结主要模式并评估稳健性。此外,为了获得更有说服力的结果,我们开发了两种调整树输入的策略,一种是通过打乱输入基因树的顺序(「--shuffle」),另一种是通过放回随机抽样树(「--引导」)。然而,排序的亚基因组在不同的重复中被随机命名。在总结结果之前,我们需要手动识别亚基因组并相应地重命名它们。该管道采用Python3语言编写,命名为「HomeoSorter」。该脚本和更多详细信息可在https://github.com/ChenRen-SCBG/HomeoSorter上获取。
HomeoSorter的模拟测试
我们首先使用模拟数据来测试新管道的性能。使用HybridSimv.1.0.1(Woodhams等人2016)总共模拟了238个数据集,这是一个可以模拟各种类型网状结构的优秀程序。更重要的是,对于每个模拟网络,HybridSim提供了一组在随机合并过程下生成的随机选择的基因树(即HybridSim定义的「合并树」),作为HomeoSorter测试的理想输入。前75个数据集用于测试经常影响网络推理准确性的两个主要因素,即父本母本贡献(即HybridSim定义的「混合分布」)和ILS。具体来说,我们模拟了具有五种不同比例的亲本贡献(50:50、40:60、30:70、20:80和10:90;从相等到不相等)和15个合并时间(CT;或「合并」)的四倍体。每个比例的比率」,由HybridSim使用,范围从0.1到200(从极高到低噪声;比率与HybridSim定义的物种形成率相关,默认为1)。每个数据集由8个分类单元和200个合并树组成。对于其他参数,我们遵循默认设置,除了关闭基因渗入,这可能会改变合并树中亲本贡献的预设比例。特别是,我们对所有模拟使用相同的种子(「-s111」)。通过这样做,HybridSim可以生成相同的网络,更重要的是, 可以减少模拟的随机性,并确保测试因素之间的差异最大程度地反映在合并树中。
接下来的15个数据集用于测试HomeoSorter在同源多倍体方面的性能。由于HybridSim无法模拟同源多倍体的系统发育,我们生成了一个简单的分叉树,并将两个姐妹类群指定为同源多倍体的两个亚基因组。同样,每个数据集都是由8个分类单元和200个合并树生成的。模拟了从0.1到200的CT,所有CT都具有相同的种子「111」。
其余数据集用于评估HomeoSorter在各种场景下的计算时间。测试了六个主要因素,包括所研究的多倍体数量、倍性水平、基因数量以及HomeoSorter的三个主要参数(「--allele」、「--best」和「--every」)。数据集使用20个分类单元和200至4000个合并树进行模拟,所有树均具有相同的「111」种子。
对于HomeoSorter分析,使用HybridSim生成的合并树作为输入。除非另有说明,否则均遵循HomeoSorter的默认设置。对于前75个数据集,我们进行了100次重复的改组分析,并总结了正确识别亲子关系的平均支持度。对于每个重复,我们进一步计算了正确等位基因分配的百分比。至于同源多倍体的测试,我们还对每个数据集进行了100次重复的改组分析,并总结了对同源多倍体模式正确识别的支持(即,四倍体的亚基因组形成一个分支,然后与其最接近的亲属成为姐妹)。然而,未评估等位基因分配,因为将等位基因分配给任一亚基因组在技术上是正确的。为了评估计算时间,我们对每个数据集进行十次重复的改组分析并计算平均时间。所有分析均在配备16核Intel Xeon E5-4657L v2 CPU(2.40GHz)和256G RAM的机器上运行。为了评估计算时间,仅使用一个线程以获得更准确的评估。
为了进行比较,我们还使用AllCoPol和Phylonet的两个模型(MPL和MPAllopp)分析了前90个数据集。上面已经提到了AllCoPol和MPL。MPAllopp(Yan等人,2022年)是一种扩展的MDC模型(Yu等人,2013年),并声称能够比Oberprieler等人(2017年)的方法更可能且更优越地处理多倍体,而无需进行排列(Yan等人,2022)。因此我们也在这里进行了测试。Allcopol测试使用默认设置运行。MPAllopp和MPL测试均在指定和不指定多倍体的情况下运行,每个测试都有20个搜索重复(「-x20」)。由于MPAllopp和MPL是为网络分析而开发的,对于同源多倍体,它只能推断出一种模式,其中导致同源多倍体的两个分支都与单个分类单元最密切相关。我们认为这是MPAllopp和MPL分析中同源多倍体的正确识别。
款冬亚族的HomeoSorter分析
对于款冬亚族的HomeoSorter分析,我们使用了Phylonet_A16分析的相同数据集,但添加了向日葵作为外群。然而,该数据集包含19个多倍体样本(款冬亚族和短舌菊属的类群),如果一起分析,将在第二轮ASTRAL分析中产生大量的亚基因组组合。由于短舌属联盟和款冬亚族亚科的主要类群都得到形态学、细胞学和分子系统发育学证据的有力支持(Nordenstam等,2009),因此我们在这里通过分析主要类群而不是物种来规避这个问题。这是通过将多倍体序列视为相应组的「等位基因」来完成的。例如,橐吾属(Ligularia Cass.)、垂头菊属(Cremanthodium Benth.)和蟹甲草属(Parasenecio W.W.Sm. & J.Small)是 LCP 复合体的成员;这三个属的所有序列在此被视为复合体的「等位基因」。还应该指出的是,两栖太平洋类群(包括黏子菊属、腋绒菊属和细叶垫菊属)在款冬亚族中的特征是其基础染色体数为x=9(很少为7),但分子系统发育证据表明,它在异源多倍体后发生分化款冬亚族(Ren等人2021)。他们也被视为六倍体谱系。该调查进行了50次改组和50次自举重复,设置相同「--等位基因8--最佳3--每3」。手动识别和重命名子基因组后,我们使用DendroPy 4.5.2版本(Sukumaran和Holder2010)的sumtrees.py脚本总结了大多数共识树和支持值(分别用于改组和引导重复)
基于同源排序的系统发育重建
为了进一步探讨HomeoSorter的实用性和缺点,我们测试了基于阶段性等位基因的系统发育重建。这里使用了50次改组重复的结果。对于每个重复,都有一个等位基因分配列表。根据每个列表,我们基于原始G727数据集构建了一个新的序列矩阵,并将款冬亚族序列重命名为其分配的亚基因组。在某些情况下,多个序列被分配给同一个亚基因组;我们生成了严格的一致序列来代表亚基因组。使用超级矩阵和多物种合并方法重建系统发育,设置与上面使用的相同。对于超级矩阵方法,为每个重复生成一个串联的ML树,并使用sumtrees.py总结多数共识树。对于合并方法,我们为每个重复的单个基因生成了ML树,然后将它们汇集在一起(总共36350个树,其中50个重复每个727个树)以总结ASTRAL-III中的物种树。
由于ASTRAL方法产生了与HomeoSorter分析类似的结果,但超级矩阵方法却没有,我们进一步测试对数据集进行过滤,以减少HomeoSorter的一些缺点的潜在影响。由于款冬亚族的单系性(包括类款冬亚族群,但不包括山绒菊属的姊妹组D亚基因组)得到了强烈支持,因此排除了那些在ML树上形成分支但分配给不同亚基因组的款冬亚族序列。然而,这种做法导致了额外的数据缺失。为了减轻它们对系统发育推断的潜在影响,我们进一步排除了款冬亚族和短舌菊属的类群样本少于15个(<75%)的基因。然后,我们使用与上述相同的设置重新运行超级矩阵和多物种聚结分析。
结果
基因组装和旁系同源
如补充表S1和图S1(Dryad)所示,除了两个DNA严重降解的样本(Capelio tabularis和Hertia cheirifolia Kuntze)外,基因恢复情况非常好,平均恢复了1040个(约94.2%)基因。对于非千里光科样品,有千里光属和1008(约91.3%)。总共854起(约77.4%)收到了Paralog警告。然而,对于具有或极有可能具有2n≤20的千里光样本以及除一个非千里光样本外的所有样本,它们有不到99个(约9%)基因接受旁系同源警告,平均有33个(约3个)%)每个样本的基因。来自异源多倍体属的狭锥花佩菊(Liu等人2013)是唯一收到大量旁系同源警告(199个基因)的非千里光科样本。相比之下,对于2n>20的千里光科样本,平均有223个(约20.2%)基因发出旁系同源警告,其范围从佛罗里达美幌菊(Arnoglossum floridanum (A. Gray) H. Rob.)的53个(约4.8%)不等。(2n=52;例如Jones1970) 到肯尼亚千里光(Dendrosenecio cheranganiensis(Cotton & Blakelock) E. B. Knox)的531个(约48.1%)(n=50;Knox和Kowal1993)中的531(约48.1%)。
对单个基因的系统发育分析表明,68个基因树中的一些部落是非单系的,这表明在部落分化之前存在潜在的基因重复。此外,309个基因存在大量缺失数据。这些基因被排除在外,以减少它们对系统发育推断的潜在影响。然后总共保留727个基因用于以下分析。
千里光族主要谱系的鉴定
基于G727_Hyb数据集的串联ML(补充图S2;Dryad)和ASTRAL(补充图S3;Dryad)分析表明菊科的部落关系几乎相同,这也与Mandel等人的结果相似。(2019)基于一组完全不同的核基因。然而,就千里光族内部的关系而言,它们之间表现出一些不一致,并且进一步不同于Pelser等人生成的部落框架。(2010)使用叶绿体基因。尽管如此,在部落内可以很容易地识别出八个主要分支/谱系,即(1)垫菊属,(2)Capelio菊科,(3)千里光亚科/Othonninae亚族(因为Othonninae总是嵌套在千里光亚科内,该分支在下文中被称为为了方便起见,称为千里光亚科(Senecioninae)),(4)短舌菊属(包括线绒菊属,短舌菊属J.R.Forst.&G.Forst,粉蟹甲属和黏菊木属),(5)山绒菊属,(6)Caputia菊科,(7)绒安菊属,和(8)款冬亚族,不包括类款冬亚族组(以下称为款冬亚族)。
在款冬亚族中,Pelser等人先前确定了五个主要类群。(2010)并且与它们的分布模式高度一致在这里得到了证实。其中包括(1)东亚类群(即LCP复合体),主要为东亚特有种,以垂头菊属、橐吾属、羽叶菊属、蟹甲草属和狗舌草属为代表;(2)中美洲群,以番蟹甲属、肉脂菊属、印第安菊属、伞蟹甲属和顶叶千里光属为代表;(3)北美西部类群,以类蟹甲属、帚蟹甲属、覆旋花属和四蟹甲属为代表,(4)温带欧亚类群,包括山雏菊属、蜂斗菜属和款冬属,(5)两栖太平洋类群,由黏子菊属、腋绒菊属和细叶垫菊属组成。根据这些结果,我们对Phylonet和HomeoSorter分析进行了二次采样。
系统网络分析
Phylonet_A1分析表明,当仅允许一种网状结构时,山绒菊属被推断为杂种(图2a)。然而,当允许有两个网状结构时,款冬亚族和绒安菊属被认为是杂交种,其亲本为款冬亚族。与山绒菊属和千里光亚科的祖先有关,绒安菊属与伞蟹甲属(中美洲款冬亚族的成员)和山绒菊属相关(图2b)。经过三项事件的测试,疆千里光属还被推断为杂种,起源于千里光亚科(图2c)。当四个网状结构被允许时,在短舌菊属谱系和千里光亚科的祖先之间推断出一个新的网状结构,并且由此产生的杂交谱系与山绒菊属谱系杂交并产生了款冬亚族。(图2d)。当允许的网纹增加到五个和六个时,与款冬亚族相关的网纹图案。和绒安菊属保持不变,但额外的多倍体千里光亚科样本甚至千里光亚科本身被推断具有杂交起源(图2e,f)。六次检验的结果对数伪似然如图2g中的柱形图所示。这些值首先从单事件测试(-1038441)快速增加到四事件测试(-1035349),然后缓慢增加到五项事件(-1036272)和六项事件(-1035247)测试,这表明四项事件的结果最适合数据。
Phylonet_A2–A15重点关注多倍体千里光亚科样本,结果表明网状结构很大程度上被推断发生在千里光亚科谱系之间(补充图S4–S17;Dryad)。在少数情况下,涉及款冬亚族样本(例如补充图S4c、S9d和S11b;Dryad),但最终产生多倍体的遗传概率均小于0.05,表明这些可能只是进化噪音。因此,千里光亚科多倍体很可能都起源于该亚族内部,与款冬亚族的起源没有直接关系。
Phylonet_A16主要针对款冬亚族(图3)。在单项事件测试中,山绒菊属仍被推断为杂种(图3a)。然而,随着更多的网状事件被允许,款冬亚族的网状图案逐渐消失。绒安菊属逐渐出现,并且比Phylonet_A1中推断的要清晰得多。在双项事件测试中,款冬亚族推测山绒菊属谱系和千里光亚科的祖先之间存在网状关系,而绒安菊属在山绒菊属和中美洲款冬亚族群的祖先之间也有另一个网状事件。(图3b)。在三项事件测试中,在千里光亚科的祖先和短舌属联盟的祖先之间推断出额外的网状结构,由此产生的杂种谱系与山绒菊属谱系杂交并产生了款冬亚族。(图3c)。款冬亚族的网状图案。尽管在款冬亚族中推断出额外的网状事件,但绒安菊属在四到六项事件测试中保持不变。(图3d-f)。至于对数伪似然值(图3g),从三项事件测试开始,它们变得总体稳定(三到六项事件测试分别为-2445021、-2444783、-2444446和-2444614)。
Phylonet_A17重点关注短舌菊属的类群(图4)。由于款冬亚族和短舌菊属的类群都被指定为杂交种,对于单项事件测试,因此推断它们具有共同的杂交起源(图4a)。当允许存在两个网状结构时,这两个群体被认为具有不同的起源,但具有与千里光亚科相关的共同亲本谱系(图4b)。对于三到六项事件测试,得到的模式通常是相似的(图4c-f)。款冬亚族推断有两个网状事件和三个亲本谱系。短声联盟仍然拥有一个亲代血统,但这一血统现在与Caputia关系密切。另外两个谱系分别与山绒菊属和千里光亚科的祖先有关。短舌菊属的类群被推断为三项事件测试(图4c)、四项事件测试(图4d)和六项事件测试(图4f)有一个网状结构,五项事件测试有两个网状结构(图4d)),但五项事件测试中的一个网状结构可以忽略不计,其边缘具有非常低的遗传概率(0.001;图4d)。这四个测试推断的亲本谱系都与Caputia菊科关系最密切(图4c-f),并且它们的对数伪似然几乎相同,在-635640到-635636之间略有波动(图4g)。
HomeoSorter的模拟测试
HomeoSorter对亲本贡献和ILS的模拟测试表明,对于亲本贡献相等(50:50)的情况(图5a),当CT=0.1(表明ILS显著)时,HomeoSorter只能在43.5时识别出亲本。%的案例。然而,随着CT增加到0.5,概率迅速增加到87.5%,并且对于CT≥1,HomeoSorter进行了所有完美的识别。等位基因分配显示出类似但更为渐进的趋势。随着CT从0.1增加到20,正确等位基因分配的百分比从约50%增加到近100%,此后保持在100%。
同样,在40:60(图5b)、30:70(图5c)和20:80(图5d)父本母本贡献的测试中,随着ILS问题变得不那么严重,HomeoSorter的性能迅速提高。然而,随着亲本贡献变得越来越不平等,亲本鉴定和等位基因分配的准确性或多或少地降低了。例如,在20:80亲本贡献测试中(图5d),即使CT≥20,HomeoSorter也无法完美识别亲本,并且等位基因分配的准确性在重复范围更广、更多的情况下显示出更大的差异。异常值(图5d)。
父本母本贡献10:90的案例有一个独特的模式(图5e)。正确识别父本母本的概率首先增加到约80%(CT=1),然后下降到50%(CT=8),此后保持在50%。同样,正确等位基因分配的平均百分比首先上升到80%(CT=6),然后下降到约50%(8≤CT≤200)。我们检查了生成的物种树,发现对于CT在8到200之间的数据集,四倍体的两个亚基因组一致成为亲本的连续姐妹,贡献主要(90%),这表明HomeoSorter接近错误识别异源多倍体称为同源多倍体。
对于同源多倍体测试,对于CT≥6的数据集,所有重复均完美恢复了同源多倍体模式(图5f)。当CT减少到4时,概率降低到96%,当CT≤2时,HomeoSorter识别失败(图5f)。
使用AllCoPol、MPL和MPAllopp对上述数据集的测试结果分别显示在补充图S18-S20(Dryad)中。在亲本比例为50:50至30:70的异源多倍体案例中,AllCoPol和MPL都表现出与HomeoSorter相似的性能,当CT≥1或2时完美识别亲子关系(补充图S18ac、S19a-f;Dryad)。然而,与HomeoSorter相比,AllCoPol在等位基因分配方面的准确性要低得多,即使对于CT≥8的病例,也只有约77%(对比接近100%)(补充图S18a-c;Dryad)。随着亲本贡献变得更加不平等,AllCoPol表现出功率显著下降(补充图S18d,e;Dryad),而MPL通常表现良好,特别是在指定多倍体时(补充图S19g-j;Dryad)。通过预先指定,即使在CR≥1时亲本贡献为10:90的情况下,亲本也能被完美识别(补充图S19j;Dryad)。关于同源多倍体测试,AllCoPol在ILS较严重的情况下完美恢复了同源多倍体模式(CTs≥0.5vs.CTs≥4;补充图S18f;Dryad),表现优于HomeoSorter,而MPL的性能相对较低。当未指定多倍体时,MPL在所有情况下都会失败(补充图S19k;Dryad)。即使有预先指定,MPL成功的案例(CR>6;补充图S19l;Dryad)也比HomeoSorter成功的案例(CR>2;图5f)要少。至于MPAllopp,在指定多倍体时通常效果很好,当CT≥1时正确识别异源多倍体(补充图S20b、d、f、h、j;Dryad),当CT≥2时正确识别同源多倍体(补充图S20l;Dryad))。然而,当没有指定多倍体时,它通常效果不佳。尽管图表上表明MPAllopp在CT≥40时成功识别出异源多倍体(补充图S20a、c、e、d、i;Dryad),在CT≥6时成功识别同源多倍体(补充图S20k;Dryad),但它应该需要注意的是,这些正确的模式只是MPAllopp同时推断出的许多(最多103个)同样简约的网络/树之一。
至于HomeoSorter的计算效率,在测试的六个因素中,时间随着五个因素(即多倍体数、倍性水平、「--等位基因」、「--最佳」和「--every」;图6a-e),但始终与基因数呈线性相关(图6f)。
款冬亚族的HomeoSorter分析
正如HomeoSorter改组分析的多数共识树所示(图7),核心千里光亚科的基本二倍体框架由三个谱系组成:山绒菊属、Caputia菊科和千里光亚科,其中后两个谱系关系更为密切。短舌菊属的类群的三个亚基因组分别附着在框架上。亚基因组A和B是Caputia的连续姐妹,都得到了全力支持。亚基因组C是大千里光亚科/Caputia菊科进化枝的姐妹,但支持相对较弱(56%/-用于改组/引导支持)。至于款冬亚族,绒安菊属有四个亚基因组和款冬亚族的五个主要类群。每个都有三个亚基因组。其中,绒安菊属的亚基因组D是独立的,与山绒菊属是姐妹,得到充分支持,而其余18个亚基因组共同形成了三个大的、强支持的进化枝(亚基因组A-C;均为100%/100%),每个进化枝均匀地由绒安菊属的一个亚基因组和款冬亚族五个主要类群的每个亚基因组组成。这三个分支分别与千里光亚科 (68%/70%)、短舌属联盟的Caputia菊科/亚基因组A和B(38%/32%)以及山绒菊属/绒安菊属亚基因组D(100%/100%)聚类。特别是,在所有这三个分支中,绒安菊属始终是中美洲款冬亚族群体的姐妹。(分别为92%/86%、100%/98%和82%/84%)。
基于同源排序的系统发育关系
我们根据排序的同源物进一步重建了系统发育。ASTRAL树(补充图S22;Dryad)与HomeoSorter改组分析的共识树大致相似,但绒安菊属的亚基因组C是款冬亚族的姐妹,而不是嵌套在其中。超级矩阵方法的共有树(补充图S23;Dryad)显示出更多差异,绒安菊属的亚基因组C和D与山绒菊属以及款冬亚族亚基因组A和B形成一个分支。
通过排除那些在单个基因树上形成分支但分配给不同亚基因组的款冬亚族序列来过滤数据集后,超矩阵(图8)和合并方法(补充图S24;Dryad)产生了相当一致的结果,即也与HomeoSorter共识树一致。如图8所示,绒安菊属的亚基因组D与山绒菊属形成了一个强支持的进化枝(超矩阵支持/局部后验概率为100%/1.0)。款冬亚族的三个亚基因组分别与短舌菊属的类群的Caputia菊科/亚基因组A和B(90%/1.0)、千里光亚科的姐妹组(98%/1.0)和山绒菊属/绒安菊属亚基因组D的姐妹组(100%/1.0)分组。特别是,在款冬亚族的三个副本中,绒安菊属始终是中美洲款冬亚族群的姐妹。具有强大的支持(均为100%/1.0)。
讨论
HomeoSorter
尽管仍处于萌芽阶段,但将等位基因定相到亲本亚基因组的策略对于异源多倍体系统发育非常有利(Oxelman等人2017;Rothfels2021)。除了解决网状模式之外,它还可以仅使用现有工具对异源多倍体进行下游分析。然而,大多数当前的等位基因定相方法依赖于简单的作图策略。例如,AlleleSorting(Šlenker等人2021)首先根据叶绿体系统发育确定所研究的多倍体的母本,然后根据多倍体等位基因与母本序列的距离将其分配给亲本亚基因组。HybPhaser(Nauheimer等人2021)构建了一个初步的系统发育主干,并将读取映射到从主干中选择的参考文献。PhyloSD(Sancho等人2022)为二倍体样本构建了系统发育框架,并通过将多倍体等位基因映射到二倍体框架来对多倍体等位基因进行定相。莱尔等人。(2023)迭代地将多倍体的共有序列与参考序列进行比较,并将位点变异分离到不同的亚基因组中以实现定相。这些作图策略虽然看似简约,但可能过于简化了复杂的异源多倍体后基因进化。特别是,他们忽视了ILS的影响, ILS是系统发育不一致的一个众所周知的原因,由于相似的不一致模式,很难将其与杂交区分开来 (Degnan和Rosenberg2009;Degnan2018),使得结果缺乏说服力。
到目前为止,最复杂的定相同源物模型是AllCoPol(Lautenschlager等人2020)和Homologizer(Freyman等人2023)。AllCoPol是Oberprieler等人(2017)方法的扩展,该方法使用排列策略和物种树重建方法将同源物分配给亲本亚基因组(更多详细信息,请参阅简介)。Lautenschlager等人(2020)通过采用启发式算法提高其效率。Homologizer是基于AlloppNET(Jones等人2013;Jones2017)开发的,这是一种贝叶斯方法,依赖于多物种合并的参数化模型和马尔可夫链蒙特卡罗算法来对跨位点的等位基因进行定相。Freyman等人(2023)通过适应高于四倍体的倍性水平、提供统计节点支持以及记录下游分析的等位基因分配来改进它。然而,AllCoPol和Homologizer都受到计算效率的限制。无论我们如何调整参数,AllCoPol都无法在具有16核IntelXeonE5-4657Lv2CPU(2.40GHz)和256GRAM的机器上运行超过180个基因(每个基因一棵树)的数据集。至于Homologizer,贝叶斯方法的一个常见问题是,对于大型数据集,分析的收敛似乎很难达到。在我们的例子中,即使对于只有100个基因的数据集,Tracerv.1.6(Rambaut等人2014)中估计的主要参数的有效样本量(ESS)在运行一个多月后通常小于30(约38000几代人)。因此我们开发了HomeoSorter。通过采用分层策略,HomeoSorter在很大程度上克服了Oberprieler等人方法的扩展问题。(2017),计算时间总是与基因数量线性相关(图6f)。基于模拟测试和我们对款冬亚族的经验案例,我们进一步证明 HomeoSorter是多倍体系统发育学的有用且可靠的工具。 下面我们更详细地讨论它的优点和注意事项。
HomeoSorter的验证。 在大多数模拟测试中,HomeoSorter都做得非常好,成功地恢复了自体/异源多倍体模式并准确地对等位基因定相(图5)。相比之下,AllCoPol在同源多倍体推断方面比HomeoSorter的性能稍好,能够在ILS较严重的情况下恢复同源多倍体模式(CTs≥0.5vs.CTs≥4;补充图S18f;Dryad)。然而,它在等位基因分配方面的准确性要低得多,即使在没有不平等亲本贡献和严重ILS问题的情况下,准确性也仅为77%左右(相对于接近100%)(补充图S18a-c;Dryad)。MPL模型在亲本贡献极其不平等(例如,10:90)的异源多倍体病例上优于HomeoSorter(补充图S19i,j;Dryad),但在ILS问题更严重的病例上表现较差(例如,失败)当CT≤6与CT≤2时;图5f,补充图S19k,l;MPAllopp在研究预先指定的多倍体时,在亲本贡献严重不平等的异源多倍体情况下也优于HomeoSorter(补充图S20h,j;Dryad)。然而,当未指定所研究的多倍体时,MPAllopp分析通常会失败(补充图S20a、c、e、g、i、k;Dryad)。此外,MPAllopp无法处理复杂的情况,例如我们的Phylonet_A1和A16数据集。这有点令人惊讶,因为它通常在简单情况下运行得非常快,但经过我们的反复测试证实了这一点。最重要的是,MPL和MPAllopp都没有实现定相等位基因的功能。
对于我们的款冬亚族经验案例,HomeoSorter似乎也正确解析了其异源多倍体模式。首先,基于改组(图7)和引导(补充图S21;Dryad)分析的HomeoSorter共识树与Phylonet分析的结果一致(图2、3)。其次,基于排序和过滤同源的共识树(图8,补充图S24;Dryad)也与HomeoSorter共识树(图7)几乎相同,并且具有更高的节点支持。第三,推断的模式得到了基本染色体数量的重要证据的有力支持(见下文)。所有这些结果有力地支持了HomeoSorter的高精度和可靠性。
HomeoSorter的注意事项。 —但是,在某些具有挑战性的情况下使用HomeoSorter时也应小心谨慎。首先,当亲本贡献极其不平等时,HomeoSorter可能无法识别异源多倍体模式。例如,在亲本贡献为10:90的情况下(图5e),即使没有严重的ILS问题,HomeoSorter也几乎将异源多倍体误认为同源多倍体,两个亚基因组始终被推断为亲本的连续姐妹(90%)贡献(CT在8-200之间;图5e)。这可能是因为ASTRAL程序倾向于支持等位基因数量更平衡的亚基因组组合。许多本应属于同一亚基因组的等位基因被分配到不同的亚基因组,导致两个亚基因组都被单独的亲本吸引。
其次,HomeoSorter的能力在一定程度上受到ILS问题的限制。HomeoSorter未能恢复所有CT≤2的同源多倍体病例和CT≤0.5的异源多倍体病例的进化模式。等位基因分配的准确性受到的影响更大,由于CT<6,其下降较早(图5)。然而,应该指出的是,这里等位基因分配的准确性可能被低估了。在生成合并树时,HybridSim还会指示等位基因的归属,并且这些指示完全被HomeoSorter接受作为评估等位基因分配的标准。对于CT≥100,合并树都与模拟网络/树一致,仅分支长度不同,并且等位基因始终是其相应亲本的姐妹。在这些情况下,实际上可以通过视觉识别多倍体模式。然而,随着ILS变得更加严重,不一致的基因树迅速增加。当CT≤6时,许多等位基因实际上与合并树上的另一个亲本关系更密切,甚至形成一个分支。在这些情况下,HomeoSorter根据遗传距离分配等位基因是合理的,但结果肯定违反了HybridSim的指示,使得分配显得不太准确。
随机性是可能影响等位基因分配的另一个问题。 这是AllCoPol(Lautenschlager等人2020)和Homologizer(Freyman等人2023)共有的常见问题。对于HomeoSorter来说,主要有三个原因。首先,这是由于HomeoSorter的分层策略,当等位基因与其他基因分组时,在每个子步骤中都会重新评估等位基因。即使等位基因最初被正确分配,这也可能改变分配。其次,尽管在测试了许多程序之后,ASTRAL程序在灵敏度、计算效率和统计一致性方面是HomeoSorter的最佳选择,但它们仍然经常无法辨别竞争等位基因/亚基因组组合之间的细微差异,并产生相同的最终归一化四重奏分数。在这种情况下,HomeoSorter将进行随机选择,从而产生随机等位基因分配。第三,复杂的进化模式使情况变得更加复杂。例如,当在第一轮ASTRAL分析中单独分析绒安菊属时,其两个亚基因组(C和D)与山绒菊属的关系为((绒安菊属_亚基因组_D,山绒菊属),绒安菊属_亚基因组_C)。当亚基因组同时存在时,很容易区分它们,但是,如果其中一个亚基因组不存在,HomeoSorter就无法区分它们,只能进行随机等位基因分配。这可能是我们基于已排序但未过滤的同源物的结果与Phylonet和HomeoSorter分析不一致的主要原因。绒安菊属亚基因组D的一定数量的等位基因可能被错误地分配给亚基因组C,导致亚基因组C被山绒菊属吸引(补充图S23;Dryad),而不是嵌套在款冬亚族的亚基因组C中。(图7、8,补充图S24;Dryad)。
附加说明 :HomeoSorter的三个重要参数(「--allele」、「-best」和「--every」)值得附加说明。「--allele」(等位基因的数量,作为将基因分成小组的标准)和「--every」(汇集在一起的基因组的数量)参数直接控制要生成的等位基因/亚基因组组合并在第一次ASTRAL分析中计算,「--best」指定在第一次ASTRAL分析中选择并传递到第二次ASTRAL分析的顶级等位基因/亚基因组组合的数量。这些参数的值越大,表明搜索越详尽,有助于准确恢复进化模式,但会增加计算时间(图6a-c),并对具有许多研究的多倍体和/或多倍体的复杂情况造成特别沉重的负担。高倍性水平(图6d,e)。因此,最好在正式分析之前调整这些参数,以在合理的计算时间和潜在的更高准确度之间取得平衡。特别值得注意的是,如果「--best」设置为「1」,HomeoSorter实际上会跳过第二个ASTRAL分析,单独分析多倍体,仅合并最后一步的结果。这正是Oberprieler等人采取的策略。(2017)和Lautenschlager等人。(2020)。然而,对于密切相关的多倍体,我们强烈建议进行第二轮ASTRAL分析。在我们的例子中,这会产生更稳定的结果。
款冬亚族的异源多倍体
通过深入的研究,我们揭示了款冬亚族的复杂起源。它包含两大类群:款冬亚族和类款冬亚族组,具有连续的异源多倍体起源。两次异源多倍体事件首先产生了现存的款冬亚族。然后,经过早期的多样化,款冬亚族的一个主要群体。参与第三次异源多倍体事件并产生类款冬亚族组。下面我们将更详细地讨论这些发现。
异源多倍体事件的数量。 ——根据所有证据,总共三个网状事件将是对款冬亚族起源最有利的情况。和类款冬亚族组。由于当添加更多网状结构时,Phylonet分析几乎总是会生成更高的对数伪似然性,因此直接确定网状结构的真实数量并不容易(Cao等人2019)。曹等人提出的实用策略。(2019)是观察所得的对数伪似然;当该值开始达到稳定水平时,相应的场景将是「正确的」场景。尽管没有在所有条件下进行充分测试(Cao等人2019),但这种策略在我们的案例中非常有意义。对于Phylonet_A1(图2d)和A16(图3c),当款冬亚族出现相同的三个网状事件时(注意这是Phylonet_A1的四事件测试,其中包括疆千里光属的额外网状),对数伪-可能性也开始达到稳定水平(图2g、3g)。后来使用更多网状结构的测试仅略微提高了该值(图2g、3g)。更重要的是,这三个网状事件自从第一次出现以来就一直被推断出来,而所有额外的网状结构都与款冬亚族的起源无关。和类款冬亚族组(图2、3)。
基础染色体数和HomeoSorter分析的证据进一步支持了这种模式。染色体证据将在下面讨论。对于HomeoSorter分析(图7),以及基于排序和过滤同源物的系统发育树(图8,补充图S24),对款冬亚族的三个亚基因组进行了排序。并分别分为三个不同的谱系。至于类款冬亚族组,有四个亚基因组,但它们只与两个祖先相关,其中一个亚基因组与亲本谱系为姐妹,其他三个亚基因组分别与第二亲本谱系的三个亚基因组聚类(图7、8,补充)图S24)。三个网状事件也是这种系统发育模式最简约的要求。
款冬亚族的异源多倍体 ——我们揭示了现存款冬亚族多样化之前的两个异源多倍体事件。第一个发生在千里光亚族的祖先和与短舌菊属联盟有关的谱系之间。然后所得的杂交谱系与山绒菊属的祖先杂交并产生款冬亚族。与千里光亚科和山绒菊属相关的亲本谱系非常清晰,正如Phylonet(图2、3)和HomeoSorter(图7、8)分析生成的网络和树上明确且一致地证明的那样。然而,与短舌菊属的类群相关的一个需要一些注释。短舌菊属的类群本身的特征还在于具有x=30的高碱基染色体数(Robinson等人,1997年;Nordenstam,2007年)。在单个基因树上,很容易观察到该联盟的多个基因拷贝,其中一个或两个与Caputia密切相关,而其他则距离较远且位置可变,表明潜在的异源多倍体起源。我们的Phylonet分析(Phylonet_A1和A16)最初没有检测到该联盟的网状信号,Caputia是它的姐妹,也是款冬亚族的亲本谱系。直接连接到它(图2、3)。当预先指定为杂种(Phylonet_A17)时,短舌菊属的类群确实通过网状事件推断出来(图4),两个亲本谱系都与Caputia最密切相关,并且其中一个亲本谱系与款冬亚族共享。(图4c-f)。在预先指定下,这显然是短舌菊属的类群的主导模式,因为不同测试之间的结果非常一致(图4c-f),并且对数伪似然值几乎相同(图4g)。然而,由于预先指定,实际上很难确定这是真实的模式还是人为的模式,因为考虑到短舌菊属的类群和Caputia之间的关系,建议的模式与Phylonet_A1的结果没有本质上的不同(图2)和A16(图3)。此外,考虑到许多个体基因树上存在与Caputia远亲相关的不同基因拷贝,网状模式可能不足以代表联盟的潜在网状历史。相反,HomeoSorter分析可以更好地总结潜在的异源多倍体模式。在这三个分选的亚基因组中,两个与Caputia和款冬亚族的亚基因组A归为一类,另一个则关系较远,但位置尚未确定(图7)。
与短舌菊属属密切相关的Caputia属使解开短舌菊属联盟起源的困难变得更加复杂。尽管具有x=10的低碱基染色体数,但基于叶绿体(cp)和核糖体(nr)基因树上的不一致位置,Caputia也被认为是一个杂种属,其中Caputia嵌套在千里光亚科内并且是姐妹分别属于短舌菊属的类群和千里光亚科远亲(Pelser等人2010)。这一假设得到了Caputia中间花微形态学的进一步支持(Nordenstam2012),这是千里光亚族分类的关键(Nordenstam等人2009)。Caputia在nrDNA树上的位置(Pelser等人2010)由我们的SLCN基因证实(图2、3、7、8)。然而,仅凭SLCN基因,实际上并没有明确的线索表明Caputia的杂交起源。在用于系统发育分析的727个基因中,只有24个基因对Caputia发出了旁系同源警告(C.medley-woodii10个,C.scaposa19个,其中5个是共享的)。此外,即使对于这24个基因,除了4个基因外,Caputia的序列都形成了一个强支持的进化枝,表明它们可能是同源等位基因,或者只是在属分化后重复。此外,Phylonet分析表明该属没有杂交信号(图2、3)。因此,短舌菊属联盟和Caputia的情况要复杂得多,我们无法对它们的进化模式提供非常有信心的答案。但对款冬亚族的结果影响并不显著。款冬亚族之间的密切关系。而短舌菊属的联盟是可以保证的,不确定性只集中在款冬亚族是否成立上。与祖先或短声联盟的祖先共享亲本血统。根据目前的证据,前一种假设更为有利。
类款冬亚族类群的异源多倍体。 类款冬亚族类群的异源多倍体模式非常简单。基于形态相似性,该类群通常被认为是款冬亚族的核心成员(Bremer1994;Robinson等人1997;Nordenstam2007)。在之前的分子系统发育研究中(Pelser等人2007,2010),它也始终是中美洲款冬亚族群的姐妹。在cpDNA和nrDNA树上,因此最初没想到它与其他款冬亚族物种有不同的起源。然而,Phylonet和HomeoSorter分析强烈表明了独立的异源多倍体起源。Phylonet一致推断类款冬亚族群体存在网状事件,其亲本与山绒菊属谱系和中美洲款冬亚族群体有关。(图2、3)。HomeoSorter对类款冬亚族群的四个亚基因组进行了分类,其中一个是山绒菊属的姐妹组,另外三个分别是中美洲款冬亚族群的三个亚基因组的姐妹。(图7、8,补充图S24;Dryad)。
染色体证据。 重要的是,推断的款冬亚族异源多倍体模式得到了基本染色体数证据的有力支持。对于款冬亚族,两个祖先山绒菊属和千里光亚科的现存最近亲,其基本染色体数x=10(Hunziker等人,1989年;Nordenstam等人,2009年)。短舌菊属的类群和Caputia与第三个祖先密切相关,其特征是x=30(例如Ornduff等人,1963;Hair1966;Beuzenberg1975)和x=10(Afzelius1967),这表明第三个祖先亲本谱系很可能也基于x=10。款冬亚族尽管有一些例外(例如,x=9,在两栖太平洋组中很少为7;Ornduff1963;Borgmann1964;Schaack等人1974),但主要基于x=30(Nordenstam等人,2009),与此处推断的异源多倍体模式高度一致。
至于类款冬亚族群体,山绒菊属代表亲本谱系之一,并且已注意到x=10(Hunziker等人,1989)。另一个亲本谱系,中美洲款冬亚族群,所有属都基于x=30(例如,Pippen1968;Turner等人1973;Robinson等人1997;Palomino等人2018),除了x=30以外。Arnoglossum为25(Jones1970),YermoDorn没有数据。因此,观察到的类款冬亚族群体的唯一碱基染色体数x=40(Turner等人,1967年;Robinson等人,1997年)与推断的异源多倍体模式完全匹配。
结论
在这项研究中,我们提出了HomeoSorter,一种用于异源多倍体系统发育研究的新同源分类管道。HomeoSorter克服了当前同源分类工具的缩放问题,并在识别多倍体模式和分配等位基因方面表现出高精度。它是异源多倍体系统发育学的可靠且急需的工具,特别是在当前的基因组时代。使用HomeoSorter结合Phylonet的MPL模型和基因组规模数据,我们为款冬亚族的起源提供了强有力的证据。我们的分析表明,款冬亚族包含两大类群: 款冬亚族和类款冬亚族组,具有连续的异源多倍体起源和相互关联的关系。 特别是,异源多倍体模式得到了基础染色体数量的重要证据的有力支持。如此清晰且有充分支持的案例在多倍体研究中很少见,并 为未来研究这种现象及其潜在的进化效应提供了极好的模型。
图表
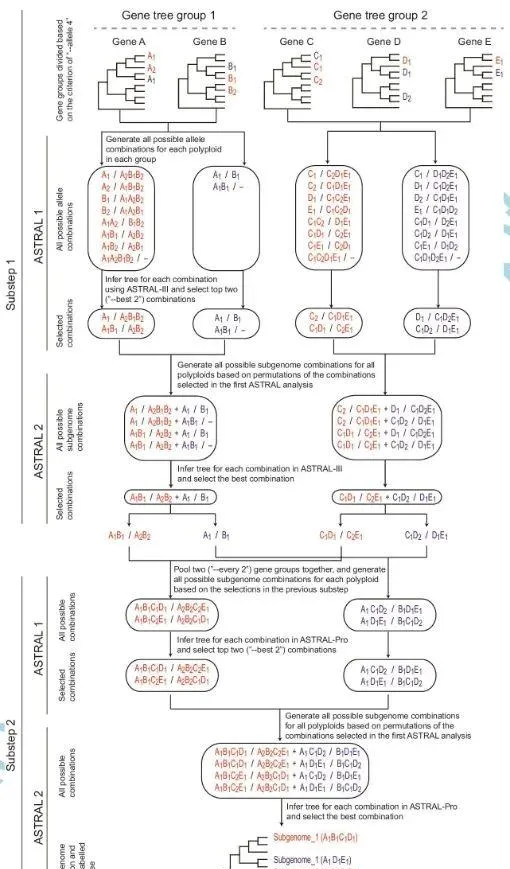
图1. HomeoSorter核心程序的工作流程,使用具有五个基因(A–E)、两个四倍体(红色和蓝色)以及参数―--allele4-best2--every2的简单案例进行说明」。A-E后面的下标表示不同的等位基因。子基因组由斜线(「/」)分隔,破折号(「–」)表示没有等位基因分配给子基因组。参数「--allele4」表明基因分组时,每组中每个多倍体的等位基因总数不能超过4个。例如,前三个基因不能组合在一起;否则,红色多倍体的等位基因总数将为六个,超出限制。参数「--best2」表明将在第一次ASTRAL分析中选择前两个等位基因/亚基因组组合,并在第二次ASTRAL分析中重新评估,「--every2」表明每两个基因组将逐渐组合从第二个子步骤开始。请注意,这个示例非常简单。整个过程仅需要两个子步骤,并且仅生成一些可能的等位基因/亚基因组组合。对于大型数据集,第二个子步骤需要重复多次,直到最终包含所有基因。此外,在更复杂的情况下(例如更多的多倍体、更高的倍性水平和/或更大的参数值),HomeoSorter将生成更多数量的等位基因/亚基因组组合。
图2. 使用最大伪似然(MPL)模型(Phylonet_A1)进行第一次Phylonet分析的结果。六次测试的结果网络分别允许一到六个网状结构,如(a)-(f)所示。橙色表示千里光亚族的样本,蓝色表示短舌菊属类群的样本,红色表示款冬亚族的样本,绿色表示Gynoxyoid组的样本。网状节点边缘附近的数字表示继承概率。柱形图(g)说明了六次测试的结果对数伪似然,表明从第四次测试开始,值开始达到稳定水平。
图3. 使用MPL模型(Phylonet_A16)进行第十六次Phylonet分析的结果。六次测试的结果网络分别允许一到六个网状结构,如(a)-(f)所示。橙色表示千里光亚族的样本,蓝色表示短舌菊属类群的样本,红色表示款冬亚族的样本,绿色表示Gynoxyoid组的样本。网状节点边缘附近的数字表示继承概率。柱形图(g)说明了六次测试的结果对数伪似然性,表明从第三次测试开始,值开始达到稳定水平。
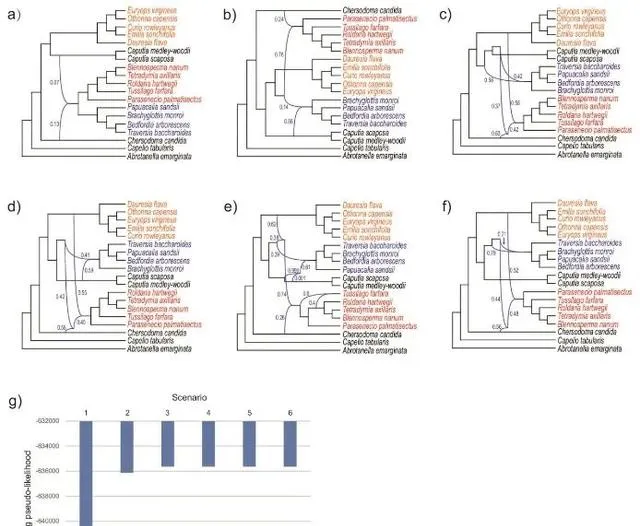
图4. 使用MPL模型(Phylonet_A17)进行第十七次Phylonet分析的结果。六次测试的结果网络分别允许一到六个网状结构,如(a)-(f)所示。橙色表示千里光亚族的样本,蓝色表示短舌菊属类群的样本,红色表示款冬亚族的样本,绿色表示Gynoxyoid组的样本。网状节点边缘附近的数字表示继承概率。柱形图(g)说明了六次测试的结果对数伪似然性,表明从第三次测试开始,值开始达到稳定水平。

图5. 基于模拟数据的HomeoSorter在不同场景下的测试结果,其中(a)-(e)为不同亲本贡献的异源多倍体,(f)为同源多倍体。折线图表示正确识别亲本(a-e)或同源多倍体模式(f)的百分比,箱线图描述了100个改组重复中正确等位基因分配的变化(a-e)。结果表明,HomeoSorter在大多数情况下都表现良好,但在谱系排序严重不完整和/或亲本贡献极其不平等的情况下,其性能会下降。
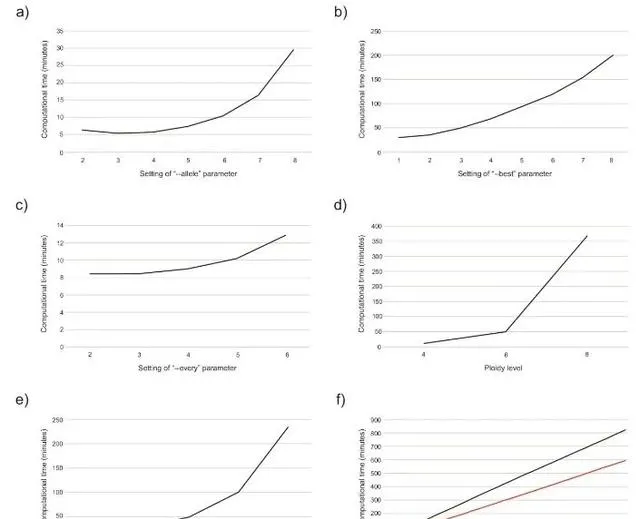
图6. 计算时间与HomeoSorter分析的六个主要因素之间的相关性。这六个因素包括参数「--等位基因」(a)、「--最佳」(b)、「-每个」(c)、倍性水平(d)、多倍体数(e)和基因编号(f)。除非另有说明,所有测试均使用单个四倍体和HomeoSorter的默认设置运行。对「--best」参数(b)的测试是用三个四倍体进行的。对于基因编号(f),测试了三种不同的场景,蓝线表示基于四倍体和参数「--allele4--best1--every2」的结果,红色表示两个四倍体,「--allele6--best3--every3」,黑色表示六倍体,「--allele6--best1--every2」。请注意,此处的计算时间是使用单个线程评估的。在实际使用中,可以采用多线程来节省时间。
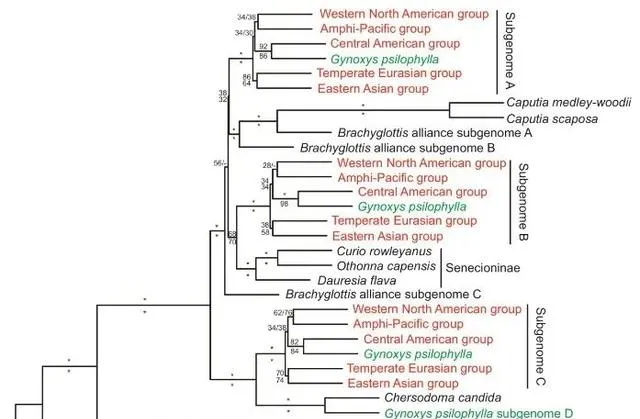
图7. 基于50次改组重复的HomeoSorter分析的多数共识树。红色表示款冬亚族的五个主要类群。绿色代表Gynoxyoid组。基于50个改组和50个引导重复总结的支持值(%)分别显示在分支的上方和下方,其中星号(「*」)表示完全支持,破折号(「–」)表示与引导分析不同的关系。
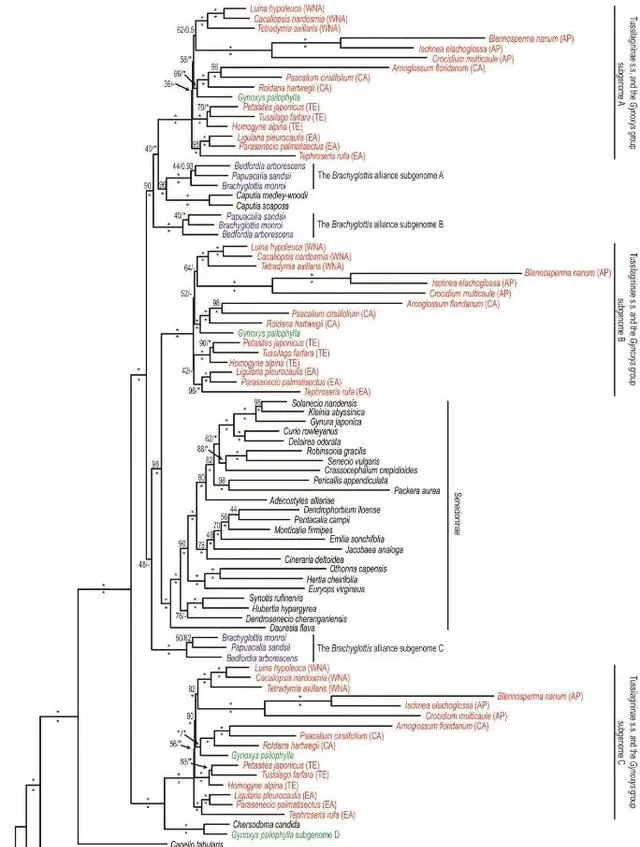
图8. 从50个超矩阵最大似然树中总结出的多数共识树,这些树是根据排序和过滤的同源物重建的。红色表示款冬亚族的样本,绿色表示Gynoxyoid组的样本,蓝色表示短舌菊属类群的样本。款冬亚族后面括号内的缩写。样本表示样本所属的五个主要类群,「AP」代表两栖太平洋类群,「CA」代表中美洲类群,「EA」代表东亚类群,「TE」代表温带欧亚类群,「WNA」代表北美西部群体。超矩阵方法的支持值(%)和合并方法的局部后验概率分别显示在分支的上方和下方,星号(「*」)表示完全支持,破折号(「–」)表明合并方法表明了不同的关系。
期刊:Systematic Biology
文章标题:Complex but Clear Allopolyploid Pattern of Subtribe Tussilagininae (Asteraceae: Senecioneae) Revealed by Robust Phylogenomic Evidence, with Development of a Novel Homeolog-Sorting Pipeline
作者信息:Chen Ren,Long Wang,Ze-Long Nie,Ming Tang,Gabriel Johnson,Hui-Tong Tan,Nian-He Xia,Jun Wen,Qin-Er Yang
原文链接:https://doi.org/10.1093/sysbio/syae046
文内图片及封面图片来源原文











