副標題:有機硼酸酯的立體選擇性亞乙烯基同系化反應合成烯基硼酸酯
同系化(homologation)反應在有機合成中具有廣泛的套用,能夠在不改變原始反應基團的情況下透過鏈延伸或環擴張直接編輯分子骨架。硼的同系化反應(即Matteson反應)透過精確控制加成順序和立體化學,在可編程或自動化有機合成中變得愈發重要。如圖1a所示,在經典的Matteson反應中碳烯以叠代方式插入硼酸酯的C-B鍵中,從而將sp3混成的碳引入分子骨架中。此外,雖然氮雜-和氧雜-Matteson反應已有報道,但將sp2混成的碳(特別是烯烴單元)插入硼酸酯C-B鍵中仍極具挑戰性。鑒於烯烴在功能有機分子骨架中普遍存在(圖1b),因此如何實作立體選擇性亞乙烯基插入以產生烯基硼酸酯就變得極為重要。目前,化學家已經報道了許多有效的方法來制備烯基硼酸酯,包括:炔烴硼氫化反應、bora-Wittig反應、Heck反應、交叉復分解等,但是從有機硼酸酯開始的合成研究卻很少。盡管使用含硼保護基的雙功能構建砌塊可以透過鈀催化的Suzuki偶聯反應實作形式的烯烴插入,但是這種方法需要硼保護和脫保護,並且與烷基硼酸酯的交叉偶聯並不是一個簡單的反應。此外,Zweifel烯化是將烯基偶聯到C-B鍵上的一種優雅方法(圖1c),不過原始硼酸酯基團在反應過程中會被消除。
近日,美國 芝加哥大學董廣彬 教授與 匹茲堡大學劉鵬 教授等研究者 透過順序和非對映選擇性插入矽烷基和烷氧基取代的carbenoids,然後進行原位立體特異性消除,便可以良好的產率和優異的 trans -選擇性將烷基和芳基硼酸酯轉化為亞乙烯基同系物 (圖1d) ,從而成功地實作了無保護基或貴金屬催化劑的「硼到硼(B-to-B)」轉化 。此外,密度泛函理論(DFT)計算揭示了carbenoid插入的非對映選擇性起源以及具有不同硫化物結合親和力的Lewis酸如何影響競爭性SN2-和SN1-型1,2-硼酸鹽遷移途徑。相關成果發表在 Nature Synthesis 上。
圖1. 亞乙烯基同系化及策略設計。圖片來源: Nat. Synth.
首先,作者選擇烷基頻哪醇硼酸酯 Int-1a 為樣版受質對反應條件進行篩選(圖2),並獲得最佳反應條件:即 Int-1a 先與大位阻鋰化烷氧基芳基硫烷試劑 S-1 (氧取代carbenoids前體)在-78℃反應得到ate-絡合物中間體,再在Lewis酸(ZnCl2)的作用下進行1,2-金屬鹽遷移並將氧取代的亞甲基插入C-B鍵中,接著在室溫下用1.2 equiv. pSO4對所得的α-烷氧基-β-甲矽烷基硼酸酯中間體進行簡單的一鍋法處理,便可以91%的產率和13:1的 E / Z 選擇性得到所需的烯基硼酸酯產物 2l 。此外,當在添加ZnCl2之前將反應混合物溫熱至室溫時,反應產率較高(90%),但 E / Z 選擇性較低(4:1)。
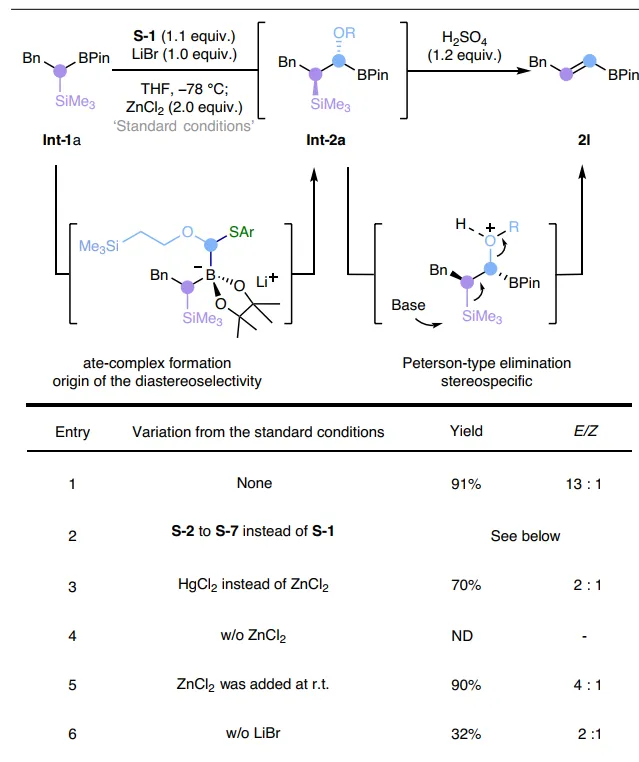
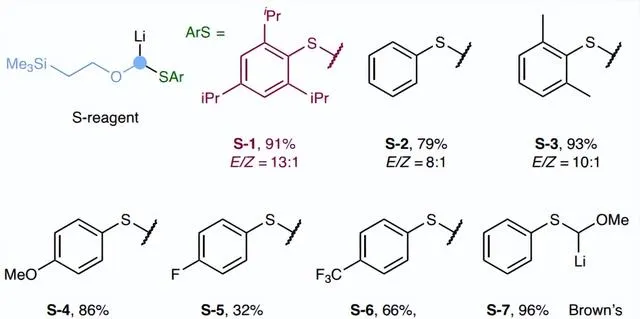
圖2. 反應條件最佳化。圖片來源: Nat. Synth.
在最優條件下,作者對烷基硼酸酯的受質範圍進行了考察(圖3),結果顯示多種伯烷基硼酸酯均能相容該反應,以良好的產率和中等至優異的 E / Z 選擇性(高達19:1)獲得相應的同系化產物,同時還能夠耐受多種官能團,包括:芳基氟化物( 2b、2i )、芳基氯化物( 2c、2j、2k )、芳基溴化物( 2d )、硫醚( 2h )等。當仲烷基硼酸酯在標準條件下進行反應時僅以較低的產率獲得所需的 E -烯烴產物,這可能是由於空間位阻增加所致。為此,作者透過使用新戊二醇衍生的硼酸酯(Bneop)來減少硼周圍的空間位阻,從而提高反應活性;同時使用位阻更大的二甲基苯基矽烷基衍生的carbenoid提供了更好的非對映選擇性,並且HgCl2的使用在一定程度上進一步提高了 E / Z 選擇性。為了簡化分離過程,作者將烯基Bneop產物直接轉化為相應的烯基碘化物。類似地,不同環尺寸的環烷基硼酸酯( 3p-3s )、帶有橋環骨架受質( 3t 、 3u )、鏈狀仲烷基硼酸酯( 3w )甚至復雜天然產物衍生的受質( 3v 、 3x )均能有效地實作這一轉化並獲得所需產物。
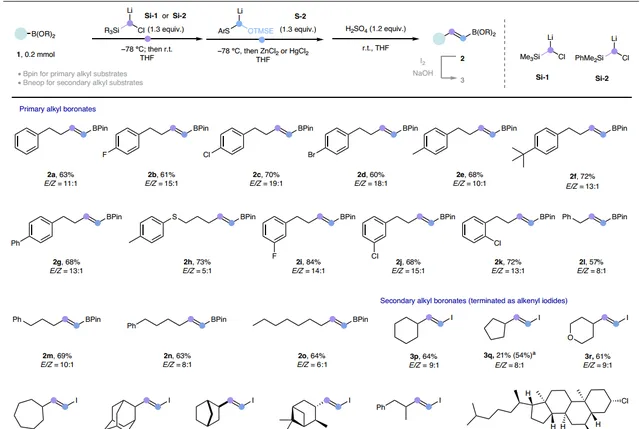
圖3. 烷基硼酸酯的受質範圍。圖片來源: Nat. Synth.
接下來,作者使用HgCl2作為Lewis酸對芳基硼酸酯的受質適用性進行了研究(圖4),結果顯示無論芳環對位、鄰位或間位帶有吸/供電子基團(如:三氟甲基醚( 5b )、硫醚( 5c )、矽烷基醚( 5d )、碘化物( 5e )、矽烷( 5f )、烷基氯( 5h )、烯基( 5i )、炔基( 5j )等),都能以良好的產率(45-76%)和優異的 E / Z 選擇性(>20:1)獲得相應產物( 5a-5t ),特別是產物 5a 能以5 mmol規模進行制備(總產率:50%)。此外,雜芳烴衍生的硼酸酯( 5u-5y )和雌酮衍生的受質( 5z )也能順利地實作這一轉化並得到同系化產物。
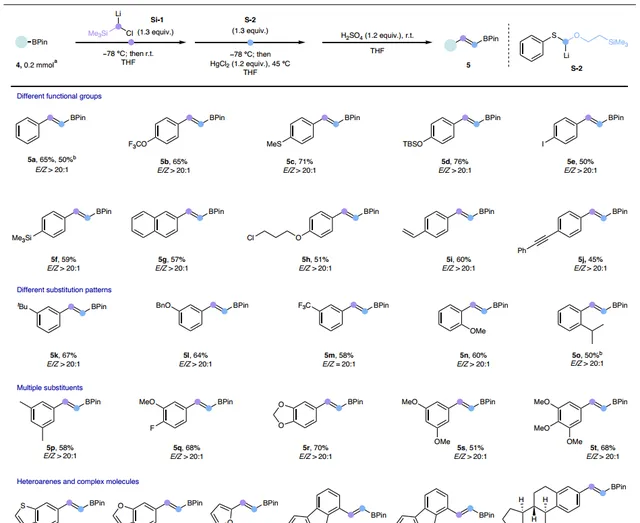
圖4. 芳基硼酸酯的受質範圍。圖片來源: Nat. Synth.
為了進一步探究反應機理,作者成功分離出α-甲氧基-β-矽烷基硼酸酯中間體( anti - 7a )並對其進行表征(圖5a),該中間體用酸處理僅產生 E -產物 5a ,進而證實了其在Peterson型消除步驟中的中間作用。為了進一步確定相對立體化學,作者將α-甲氧基-β-矽烷基硼酸酯中間體( anti - 7b )進行亞甲基同系化反應,然後經氧化和p-nosyl保護便可得到亞磺酸酯 8 ,其X-射線繞射分析證實矽基和甲氧基處於 anti -構型。其次,作者還透過DFT計算來研究carbenoid插入步驟的非對映選擇性和Lewis酸效應。如圖5b所示,烷氧基取代碳負離子 11 與α-甲矽烷基硼酸酯 10 加成過程的計算表明經過渡態 TS1a 形成的硼酸酯 12a 是動力學上有利的產物;而另一個非對映異構體 12b 則由於芳基硫醇鹽LG和硼酸酯 10 上α-甲基之間的不利交互作用導致過渡態 TS1b 能壘比 TS1a 高1.3 kcal mol-1。隨後,作者探索了ZnCl2的Lewis酸效應(圖5c),其中ZnCl2傾向於與芳硫基化物的硫原子和硼酸酯的氧原子結合並形成 13a 。從 13a 開始,經協同過渡態 TS2 進行的SN2型途徑是最有利的,活化自由能(Δ G ‡)為15.6 kcal mol-1;而SN1型途徑涉及逐步LG解離形成 14 (吸熱),然後透過 TS3a 或 TS3b 進行1,2-遷移,因此不太有利。由於立體特異性SN2型1,2-遷移( TS2 )會導致烷氧基取代碳中心的立體化學完全反轉,因此該過程將 13a 轉化為α-甲氧基-β-矽烷基硼酸酯 15 的 anti -非對映異構體,再在立體特異性Peterson消除後選擇性地形成 E -烯烴,這與實驗觀察到的立體選擇性相一致。鑒於汞比鋅具有更強的硫醇鹽結合親和力,因此當使用HgCl2作為Lewis酸時,非立體特異性SN1型1,2-遷移途徑可能更有利。事實上,HgCl2促進芳基硫醇鹽解離的Δ G ‡僅為3.3 kcal mol-1,比類似ZnCl2介導LG解離過程的能壘低12.2 kcal mol-1(圖5d)。另外,由於中間體 14 經 TS3a 和 TS3b 進行1,2-遷移的能壘差較小,因此使用更親硫的Lewis酸HgCl2時逐步SN1型途徑更有利,從而導致同系化產物的非對映選擇性降低。

圖5. 機理研究。圖片來源: Nat. Synth.
此外,作者探索了同系化反應的合成套用。具體而言:1)該方法可用於生成掩蔽的烯烴單元,可以耐受氫化條件(圖6a);2)當使用4.0 equiv ZnCl2時,無需汞鹽即可實作芳基硼酸酯的同系化反應(圖6b),並獲得單一的 E -異構體;3)該方法的合成潛力在piperdardine家族天然產物的簡化合成中得到了進一步證明(圖6c)。從共同受質芳基硼酸酯 4r 出發,透過一鍋法雙亞甲基同系化、亞乙烯基同系化和Suzuki反應便可以三步、32%的總產率、12:1 E / Z 選擇性合成piperdardine(Route A),並且該過程僅需一次純化。類似地,作者透過Route B制備piperdardine類似物,總產率為67%。為了制備三烯天然產物retrofractamide A,作者透過亞乙烯基同系化、雙亞甲基插入、亞乙烯基同系化和Suzuki反應實作其合成(Route C)。與先前的方法相比,這種叠代合成策略步驟更少、總產率更高,最大限度地減少反應中間體的純化,更重要的是為合成設計提供了可編程性。
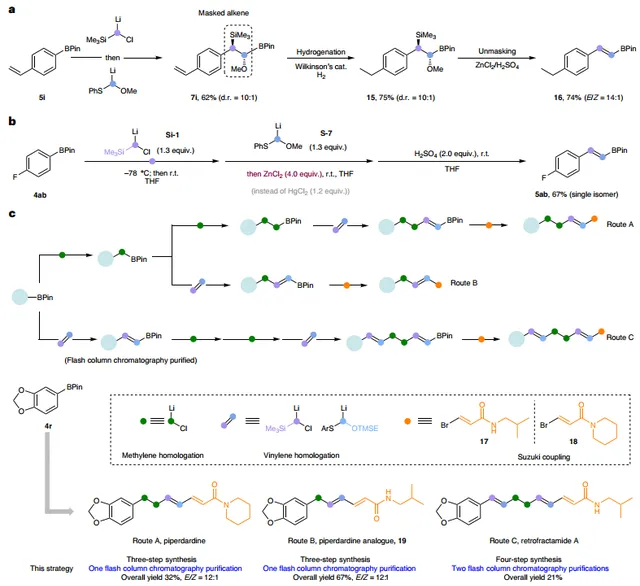
圖6. 合成套用。圖片來源: Nat. Synth.
鑒於ate-絡合物(如: 9a )的形成是非對映選擇性的,並且軟Lewis酸(如:ZnCl2和HgCl2)在1,2-金屬鹽遷移過程中促進芳基硫醇鹽作為LG,因此用親氧(即「硬」)Lewis酸(如:AlCl3或CeCl3)代替軟Lewis酸就可以將烷氧基作為LG,從而選擇性地獲得中間體 syn - 20a (>15:1 Z / E ,圖7a),比α-甲氧基硼酸酯 7a 具有更強的穩定性,可進行快速柱色譜純化和光譜表征。為了有效地促進 anti -消除,作者發現對位氯取代的芳基硫醇鹽( S-8 )效果更好。當用1.2 equiv TBAF和1.5 equiv甲基乙烯基碸處理中間體 20 時,能以良好的產率和中等的非對映選擇性獲得所需的 Z -烯基硼酸酯( 21a-f ),其中非對映選擇性的降低可能是由於消除步驟中芳基硫醇鹽LG引起的差向異構化所致。
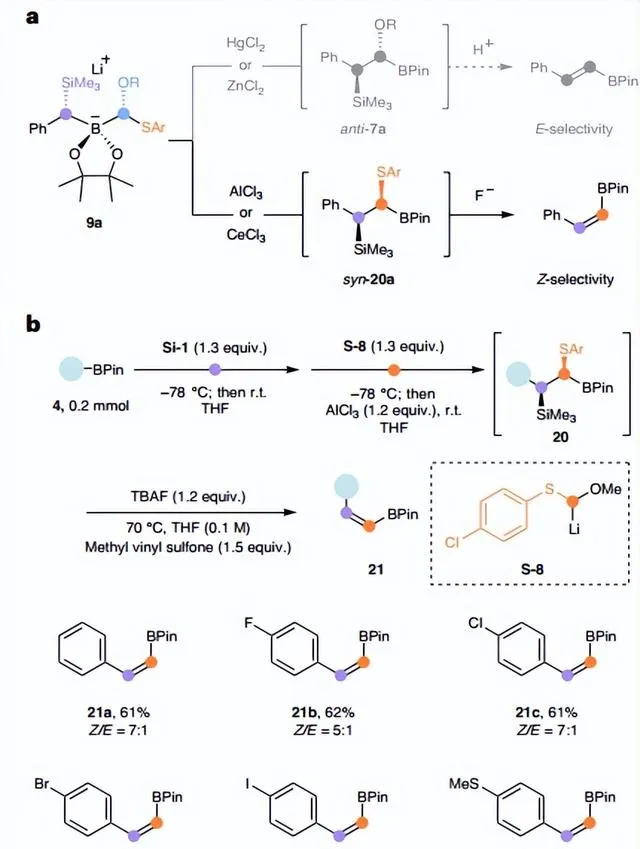
圖7. 芳基硼酸酯的 Z -亞乙烯基同系化。圖片來源: Nat. Synth.
總結
董廣彬課題組和劉鵬課題組透過順序和非對映選擇性插入矽烷基和烷氧基取代的carbenoids,然後進行原位立體特異性消除,便可以良好的產率和優異的 trans -選擇性將烷基和芳基硼酸酯轉化為亞乙烯基同系物,甚至可以透過叠代亞乙烯基和亞甲基同系化反應來實作piperdardine家族天然產物的可編程合成。此外,DFT計算揭示了carbenoid插入的非對映選擇性起源以及具有不同硫化物結合親和力的Lewis酸如何影響競爭性SN2-和SN1-型1,2-硼酸鹽遷移途徑。
Synthesis of alkenyl boronates through stereoselective vinylene homologation of organoboronates
Miao Chen , Thomas H. Tugwell, Peng Liu, Guangbin Dong
Nat. Synth ., 2024 , DOI: 10.1038/s44160-023-00461-w











