啟:成功的花朵,是用努力的汗水澆灌出來的,感謝閱讀本篇文章
文字| 洛索祥子
編輯| 洛索祥子
前言
BIAN型配體是一類具有豐富配位和氧化還原化學性質的活性芳香族受體二亞胺。
這些配體還含有一個中心1,4-二氮雜丁二烯片段,我們可以用萘骨架對其進行補充,這種結構使得BIAN型配體同時具有較強的σ-供體性質和π-受體性質,從而保證了金屬離子在配體中的穩定性。高氧化態和低氧化態。
由於這種過渡金屬配合物在催化、材料科學和生物化學等領域發揮著不可替代的作用,我們決定透過X射線繞射對其進行分析,分析配體與金屬之間的配位模式以及分子幾何結構。 配合物在不同氧化還原態之間的轉變可用於提高我們對還原化學的理解。
X射線結構描述
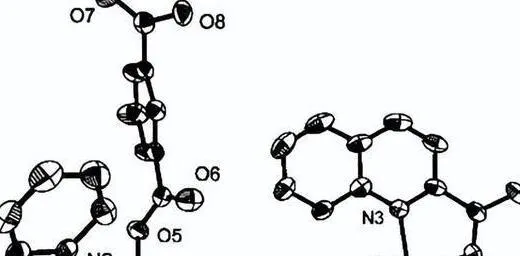
適合於X射線繞射分析的單晶是透過從二氯甲烷/甲苯混合物中緩慢蒸發獲得的,而適合於X射線繞射分析的2和3的單晶分別是從二氯甲烷/己烷(2)和二氯甲烷中獲得的。 甲烷/乙醚,(3)溶劑混合物。
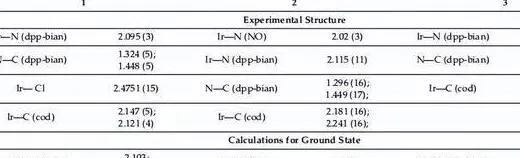
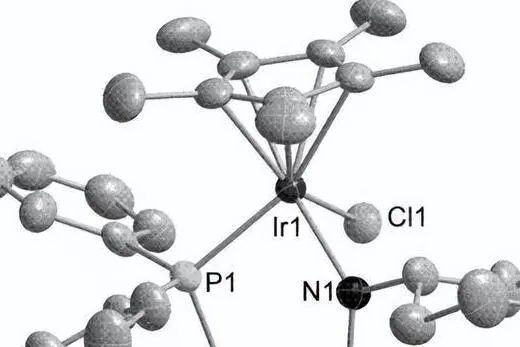
配合物1具有扭曲四角錐結構的配位中心,銥的配位環境包括兩個dpp-bian氮原子、兩個環辛二烯π鍵的中點和一個軸向氯原子。 我們需要註意,銥-氯鍵 (2.480 A) 比銠類似物 [Rh(cod)(dpp-bian) 中的銥-氯鍵 (Rh-Cl,2.580 A) 稍長,但短 (0.1 A) )Cl]。
在配位的dpp-bian中,CC鍵(1.458 A)稍微縮短,而C = N鍵(1.314 A)與[Rh(cod)(dpp-bian)Cl]中的鍵相同(CC, 1.486 A 和 C = N, 1.294 A),這可能表明二亞胺片段中的電子密度更具共振性。 此時的Ir-N鍵長為2.080 A,處於類似配合物的Ir-N鍵長範圍內。 [48-50]。
同時,配合物2也有類似的結構; NO基團的氮原子位於四角錐頂而不是氯原子,外配位球含有兩個BF4陰離子,其中二亞胺基團中的CC和C=N,鍵長分別為1.50A和1.30分別為A,表示dpp-bian的中性狀態。
Ir-N (dpp-bian)鍵長為2.12 A,略長於配合物1,Ir-N (NO)距離為1.95 A,在硝酰基銥配合物中觀察到的Ir-N鍵長範圍內。 Ir-NO角為125o,NO鍵長為1.2 A,是銥硝酰配合物中典型的彎曲硝酰基。
在配合物3中,銥的配位環境具有由兩個dpp-bian氮原子和兩個環辛二烯π鍵組成的方形幾何結構。 dpp-bian 中的 CC 和 C = N 鍵長分別為 1.493 A。 和1.298 A,對應於配體的中性狀態。
這導致帶正電的復合陽離子之間形成二聚體,但這些二聚體也形成晶體包,其中陰離子和溶劑分子填充自由空間。
在2和3的晶體堆積中,可以觀察到晶體具有三個贗通道。 該二聚體不存在於1的晶體結構中,這可能是由於絡合物的中性電荷所致。
為了研究晶體結構2和3中的分子間π-π交互作用,我們進行了DFT計算,並使用ωB97XD/DZP-DKH理論對模型超分子配合物中的電子密度分布進行了拓撲分析。 等級。
隨後,利用QTAIM方法對模型超分子配合物中的電子密度分布進行拓撲分析,發現晶體結構2和3中各種分子間接觸的鍵臨界點的存在。
這些分子間接觸的電子密度低振幅(0.003-0.006 au)、正電子密度拉普拉斯值(0.009-0.018 au)以及這些分子間C·C接觸的鍵臨界點處的能量密度為正零或非常接近零的值(0.000–0.001 au),以及它們的估計強度(0.3–0.6 kcal/mol),是類似化學系統中典型的 π-π 和相關交互作用。
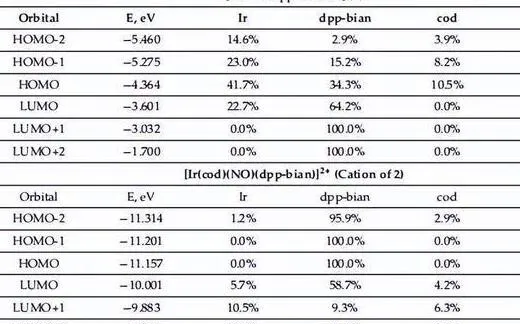
在晶體結構2和3中,分子間C·C接觸鍵臨界點處的動能密度G和勢能密度V之間的平衡(即-G/V > 1)表明這些交互作用是純粹的非共價[61],負的λ2值證實了這些接觸的重力性質。
拉普拉斯電子密度分布、鍵合路徑和選定的零通量表面的等高線圖; 對晶體結構 2 和 3 –C 接觸中選定的分子間 C 進行電子定位函式 (ELF) 和降低密度梯度 (RDG) 分析。
氧化還原特性
在CpCl2中,我們利用迴圈伏安法(CV)研究了銥配合物1和3的氧化還原性質,溶液1的迴圈伏安圖顯示出兩個準可逆還原波,分別位於E1/2 = -0.30 V處(E = 110 mV) 和 E1/2 = -1.15 V (E = 110 mV),以及 E1/2 = 0.60 V (E = 96 mV) 時的準可逆氧化過程。
類似地,在銠(I)、鈀(II)或鉑(II)與dpp-bian的絡合物的CV中也檢測到兩個特征可逆還原波[17,38]。 這些過程被認為是以配體為中心的,對應於 BIAN 配體的連續雙電子還原,形成 BIAN 單陰離子和 BIAN 二陰離子。
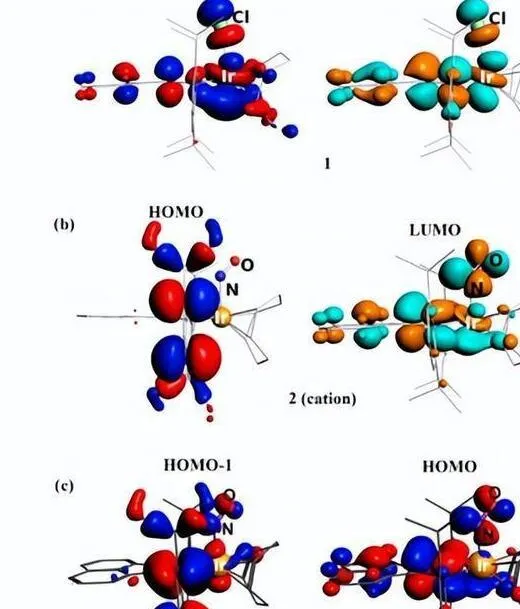
然後1的DFT計算結果證實了dpp-bian對最低未占據分子軌域(MO)(64%)的主要貢獻,盡管銥軌域對LUMO(23%)的貢獻也很大。
相反,氧化過程可能涉及金屬中心的 IrI/IrII 氧化還原對,然而,1 的最高占據分子軌域 (HOMO) 僅由 42% 的銥軌域組成,dpp-bian 的貢獻顯著 (34% ),因此,這種氧化過程更稱為混合金屬/配體中心的氧化。 值得註意的是,氧化過程是可逆的(準可逆),這表明氧化產物具有一定的穩定性。
此外,配合物3具有類似的還原模式,在E1/2 = -0.20 V(E = 83 mV)和E1/2 = -1.14 V(E = 70 mV)處有兩個準可逆還原波,並且我們得到了不可逆氧化還檢測到Ea = 1.55 V,與1相比,有明顯的陽極位移,對應的再還原峰位於Ec = 1.16 V。根據DFT計算,3的HOMO和LUMO完全定位分別在 Ec = 1.16 V 時。 Ir (98%) 和 dpp-bian (99%)。
因此3的氧化可以看作是一個純金屬中心的過程,與1相比是不可逆的。同時,BIAN配體參與氧化過程是保證其可逆性的關鍵因素。 對於銠類似物 [Rh(cod )(dpp-bian)]+,已報道了不可逆的金屬中心氧化。
不僅如此,我們還發現配合物1和3的氧化還原過程的陽極和陰極峰值電位幾乎與電位掃描速率(50-200 mV/s)無關,表明這是一個電化學可逆過程,並且峰值電流與掃描速率的平方根的比率,即I·ν^(-1/2)與掃描速率,保持恒定,這是擴散控制電子轉移反應的特征。
dpp-bian 的自由基陰離子狀態
正如我們之前指出的,1中dpp-bian配體內的CC和C=N距離不符合中性態dpp-bian的典型特征。 與 2、3 和 [Rh(cod)(dpp-bian)Cl] 的情況不同,這些值更適合 dpp-bian 的自由基陰離子狀態而不是中性狀態。
FT-IR 光譜中 ν (C = N) 和 ν (CC) 振動帶的位置與 dpp-bian 上的過剩電子密度一致。 dpp-bian 自由基陰離子的形成可以透過電子密度從銥 (I) 轉移到配體來解釋,在這種情況下,配合物 1 應被視為具有兩個不成對電子(一個在銥上,一個順磁性 [IrII(cod) )(dpp-biano/-)Cl] 在 dpp-bian 上)。
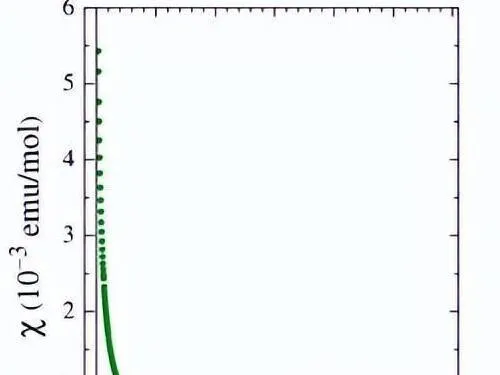
然而,磁化率測量表明 1 在高達 20 K 的溫度下表現出抗磁行為,低於該溫度則存在微弱的順磁性鐵雜質貢獻(~0.1 at.%)。 這種明顯缺乏順磁響應表明 [IrI(cod)(dpp-bian0)Cl] 的單重態 (S = 0)。
另一方面,對 1 進行的 DFT 計算排除了從基單重態到激發順磁態的躍遷,因為發現反磁態比激發順磁態更有利,能量差為 81 kJ/mol,然而,計算得出的[Rh(cod)(dpp-bian)Cl]中銥原子的電荷略高於銠原子的電荷,而dpp-bian上有少量負電荷,這可能表明電子金屬和dpp-bian配體之間的密度變化它們之間存在高度共振。
基於這些結果,我們認為,盡管電子密度的損失會影響 C=N 和 CC 距離以及相應的振動頻率,但配合物 1 中 dpp-bian 的電荷狀態在形式上最好描述為中性。
我們還可以看到 1 的可逆氧化促使我們嘗試透過合適的氧化劑(本例中為 NOBF4)對 1 進行單電子氧化,以制備順磁性 Ir(II) 配合物 [IrII(cod) (dpp-bian)然而,Cl]+ 1 與 NOBF4 的反應導致形成亞硝基絡合物 2。
不仔細看,這個反應可以描述為非等電交換,用NO+取代Cl-,維持Ir(I)的氧化態,形成IrI(cod)(NO+)(dpp-bian)2,其中大部份已知銥亞硝酰配合物均被描述為含有NO+基團,並且我們還可以假設NO+與Ir(I)氧化加成,形成IrIII(cod)(NO-)(dpp-bian)2。
有一種假設認為,彎曲的NO構型(2中Ir-NO角為125°)表明NO上帶有負電荷,甚至有報道稱含有彎曲亞硝基配體的銥(III)方錐體絡合物。 然而,[Ir(NO)(SH)2(PPp)2],人們必須意識到這種方法的局限性。
NO鍵主要是共價鍵,取決於{M-NO}單元的電子總數、自旋局域化和前沿軌域中π鍵的數量。 在不改變MNO角度的情況下可以獲得金屬和NO的形式電荷。 計算得到的2Ir-NO角和其他幾何參數與X射線繞射數據吻合良好。

因此,配合物2的基態可以正式解釋為抗磁性(單態)IrI(cod)(NO+)(dpp-bian)2或IrIII(cod)(NO-)(dpp-bian)2。
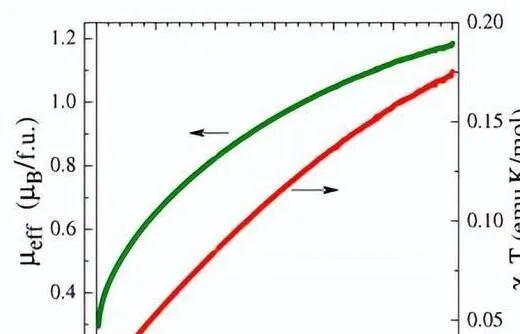
在 2 的靜態磁測量中,磁矩隨著溫度的升高而增加,直到達到 1.2 uB(在 300 K 時),表明存在順磁態,因此除了 2 的基單重態之外,人們預計還存在激發態三重態。
鑒於 2 中存在三個可以改變其價態的中心(Ir、NO 和 dpp-bian),這可能是分子內的電荷轉移轉變(氧化還原異構體轉變),或者僅僅是 2 Singlet 的結構片段內-三重態躍遷,沒有顯著的電荷重新分配。
氧化還原異構轉變中可能的選擇之一是電子從 NO 基團轉移到 Ir(III),形成 IrII(cod)(NO0)(dpp-bian)2 的兩個順磁中心:d7-Ir (II) 離子和對於 NO0 組,單線態-三線態配體內過渡選項似乎更現實。
由於 dpp-bian 對 [Ir(cod)(NO)(dpp-bian)]2+(2 的陽離子)的前沿分子軌域做出了重大貢獻,計算出單重態和三重態之間的能量差(113.0 kJ/mol) 甚至高於 1。
透過 EPR 光譜,我們在 2 的溶液中檢測到 77K 的順磁性物質,然而,這種 ESR 活性物質是一種完全不同的復合物,S = 1/2,並且一定是分解產物,考慮到亞硝酸鹽的不穩定性復合物 2 的 NO 基團,我們提出了 2 分解的兩種可能途徑。
第一種方法涉及從 2 中消除 NO+(作為 NOBF4)並形成 Ir(I)、IrI(cod)(dpp-bian) (3) 的非磁性配合物,另一種可能性是消除 NO 瓦斯形成S=1/2的順磁配合物IrII(cod)(dpp-bian)2(4),它是溶液中的ESR活性物質。
事實上,目視觀察到單晶 2 的破裂伴隨著瓦斯(可能是 NO)的釋放,我們計算出 Ir-NO 鍵解離能為 173–179 kJ/mol,要麽是透過消除 NO+ 要麽是NO 的釋放,遠小於計算的 Ir-Cl 鍵解離能(447 kJ/mol),證實了 NO 基團的容易消除。
此外,計算出的4的ESR參數與實驗數據吻合良好,因此EPR光譜數據可以與溶液中2的分解相結合,形成Ir(II)配合物IrII(cod)(dpp-bian)2 (4) 然而,我們試圖將這種順磁復合物分離為單獨的相,但沒有成功,這顯然是由於其穩定性較低。
這與 CV 數據間接一致,因為 CV 數據表明 3 的不可逆氧化,因此 4 作為 3 的單電子氧化產物的不穩定性。
總結
在本實驗中,我們獲得了一系列新的銥配合物,即[Ir(cod)(dpp-bian)Cl] (1) Ir(cod)(NO+)(dpp-bian)2, (2)和Ir(cod) )(dpp-bian), (3) 這些配合物具有龐大的氧化還原活性 dpp-bian 配體。
同時,配合物2是銥配合物中罕見的亞硝基配合物,其亞硝基配體具有彎曲結構。 關於配合物1和2,我們發現銥和配體(dpp-bian、亞硝基)處於電荷態,這是由於DPP-bian和亞硝基配體的無害行為。
因此,配合物1和3表現出金屬/BIAN配合物典型的可逆兩步、雙電子還原反應是相對正常的。
此外,我們還檢測了1的可逆混合金屬/配體中心氧化反應,並透過靜磁化率方法研究了2在1.77~300 K範圍內的磁效能。 隨著溫度升高,磁矩也增加至 1.2 uB(300 K 時)。
這種磁性行為可以透過熵驅動的熱誘導氧化還原異構化過程來解釋,但變溫光譜研究和DFT計算並未證實這一假設,導致2的磁性行為的本質仍未得到解決。
參考文獻[1]【基於活性氧化還原BIAN的二亞胺配體在金屬催化小分子合成中的套用】。
【2】「含有無害配體的不同大小的螯合環」。
【3】「第5族金屬與二亞胺配體的單核和雙核配合物:合成、反應性和套用前景」。
[4]「具有氧化還原活性雙胺基配體的 Bisalane:對炔烴的獨特反應性」。
【5】「鎂、鈣、銪、鎵、鋅與雙(亞胺基)苊配體配合物的合成與結構」。
幕:只有經歷過挫折和磨難,我們才能真正理解成功的價值,謝謝觀看











