
本文約4000字,建議閱讀8分鐘平均絕對誤差僅 2.2 meV。
清華大學研究人員利用原創的深度學習密度泛函理論哈密頓量 (DeepH) 方法,發展出 DeepH 通用材料模型,並展示了一種構建「材料大模型」的可行方案,這一突破性進展為創新材料發現提供了新機遇。
在材料設計中,了解其電子結構與性質是預測材料效能、發現新材料、最佳化材料效能的關鍵。過去, 業界廣泛使用密度泛函理論 (DFT) 來研究材料電子結構和性質,其實質是將電子密度作為分子(原子)基態中所有資訊的載體 ,而不是單個電子的波函式,從而將多電子體系轉化為單電子問題進行求解,既簡化了計算過程,又可以確保計算精度,能更準確地反映孔徑分布。
然而,DFT 的計算成本極高,通常只能用於研究小尺寸的材料系統。受到材料基因組倡議的啟發,科學家們開始嘗試利用 DFT 構建龐大的材料資料庫,雖然目前只收集到了有限的數據集,但這已經是一個了不起的開始。以此為開端,隨著 AI 技術帶來的全新變革,研究人員開始思考,「將深度學習與 DFT 進行結合,讓神經網路深入學習 DFT 的精髓,能否帶來一場革命性突破?」
這正是深度學習密度泛函理論哈密頓量 (DeepH) 方法的核心。 透過將 DFT 的復雜性封裝在一個神經網路中,DeepH 不僅能夠以前所未有的速度和效率進行計算,而且隨著訓練數據的增加,其智慧也在不斷提升。 近日,來自清華大學物理系的徐勇、段文暉研究組成功利用其原創的 DeepH 方法,發展出 DeepH 通用材料模型,並展示了一種構建「材料大模型」的可行方案,這一突破性進展為創新材料發現提供了新機遇。
相關研究以「Universal materials model of deep-learning density functional theory Hamiltonian」為題,已發表於 Science Bulletin。

論文地址:
https://doi.org/10.1016/j.scib.2024.06.011
開源計畫「awesome-ai4s」匯集了百余篇 AI4S 論文解讀,並提供海量數據集與工具:
https://github.com/hyperai/awesome-ai4s
透過 AiiDA 構建大型材料資料庫,針對性排除磁性材料幹擾
為了證明 DeepH 通用材料模型的普適性,該研究透過自動互動式基礎設施和資料庫 (AiiDA) 構建了一個包含 104 種固體材料的大型材料資料庫。
為了展示多樣化的元素組成,該研究還選擇了元素周期表的前四行,從而排除掉了從 Sc 到 Ni 的過渡元素,以避免磁性材料幹擾,並排除了稀有瓦斯元素。候選材料結構則來源於 Materials Project 的資料庫。除了基於元素型別進行過濾之外,候選材料在材料計畫中被進一步細化為只包括那些標有「非磁性」的材料。為簡單起見,在晶胞中包含超過 150 個原子的結構被排除在外。
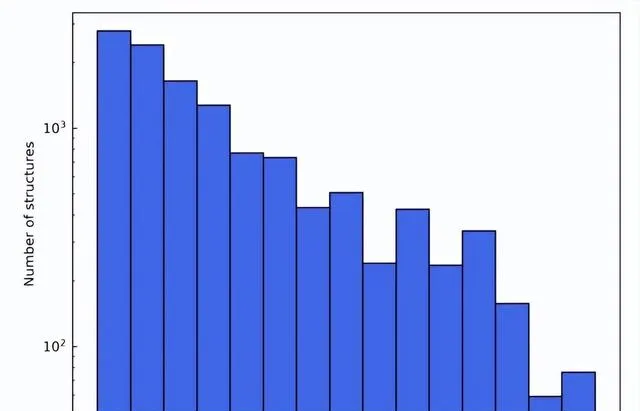
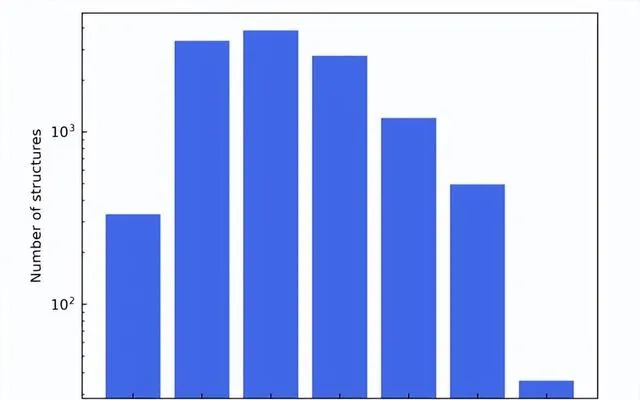
通用數據集的每個結構中原子和元質數量的分布
作為這些過濾標準的結果,最終的材料數據集由總共 12,062 個結構組成。在訓練過程中,數據集按 6:2:2 的比例劃分為訓練集、驗證集和測試集。接下來, 該研究利用 AiiDA(自動化互動式基礎設施和資料庫)的框架開發了一個高通量的工作流程來進行密度泛函理論計算,並用它來構建材料資料庫。
以 DFT 哈密頓量為目標,用 DeepH-2 方法訓練 DeepH
研究認為, DFT 哈密頓量 (DFT Hamiltonian) 是理想的機器學習目標。
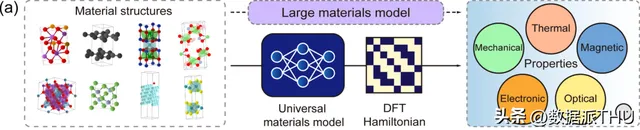
開發能夠描述材料結構-性質關系的大型材料模型的可行途徑
首先, DFT 哈密頓量是可以直接從總能量 (total energy)、電荷密度 (charge density)、能帶結構 (band structure)、物理響應 (physical responses) 等物理量中匯出的基本量, DeepH 通用材料模型則可以接受任意材料結構作為輸入,並生成相應的 DFT 哈密頓量,從而可以直接推導各種材料特性,如上圖所示。
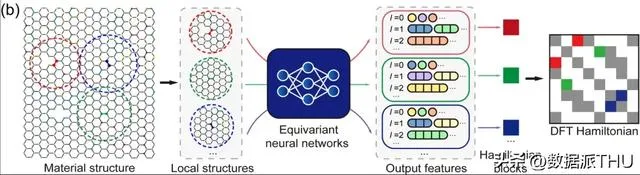
DeepH 根據局部結構資訊分別學習和預測 DFT 哈密頓矩陣塊的工作原理
其次, 在局域原子基組下,DFT 哈密頓量可以表示為稀疏矩陣,其矩陣元由局部化學環境決定。 在等變神經網路 (Equivariant neural networks) 中,DeepH 利用不同角量子數 l 標記的輸出特征來表示 DFT 哈密頓量,如上圖所示。因此,人們可以根據臨近結構資訊來建模原子對之間的哈密頓量矩陣元,而不需要對整個材料結構的 DFT 哈密頓量矩陣進行建模。這不僅大大簡化了深度學習任務,而且極大地增加了訓練數據量。在推理方面,一旦深度學習網路學習到足夠多的訓練數據,經過訓練的模型就可以很好地推廣到更多未被見過的新材料結構。
DeepH 的關鍵思想是利用神經網路來表示 HDFT。 透過改變輸入的物質結構,首先建立的是由 DFT 程式碼生成的 HDFT 訓練數據,然後將這些數據用於訓練神經網路。這些經過訓練的網路模型隨後再被用來對新的物質結構進行推理。
在這個過程中,存在兩個非常重要的先驗知識——其一是局部性原則, 該研究在局域原子樣本中表示 DFT 哈密頓量,並將哈密頓量分解為描述原子間耦合或原子內耦合的塊。因此,單個訓練材料結構可能對應於大量數據的哈密頓量塊。此外,每個哈密頓量塊可以根據局部結構的資訊而不是整個結構來確定。這種簡化確保了 DeepH 模型的高精度和可轉移性。
其二是對稱性原理, 當從不同的座標系觀察時,物理定律保持不變。因此,相應的物理量和方程式在座標變換下表現出等價性。保持等價性不僅提高了數據效率,而且增強了泛化能力,這可以顯著提高 DeepH 的效能。第一代 DeepH 架構透過局部座標系簡化了等價問題,並透過局部座標的變換恢復了等價特征。第二代 DeepH 架構基於等價神經網路,名為 DeepH-E3。在此框架中,所有輸入、隱藏和輸出層的特征向量都是等價向量。最近,這項工作的作者之一提出了深度學習的新一代架構 DeepH-2。在效率和準確性方面,DeepH-2 表現最優。
綜上,該研究的深度學習模型 DeepH 使用 DeepH-2 方法訓練,共包含 1,728 萬個參數,基於 3 個等價變換塊組成了可用於訊息傳遞的神經網路,每個節點和邊緣攜帶 80 個等價特征。 材料結構的嵌入包含原子序數和原子間距離,采用高斯平滑策略,基函式的中心範圍從 0.0 一直到 9.0Å。神經網路的輸出特征則透過線性層傳遞,然後透過 Wigner-Eckart 層構建 DFT 哈密頓量。
該研究在 NVIDIA A100 GPU 上進行訓練,共進行了 343 個時期,耗時 207 小時。在整個訓練過程中,batch 大小固定為 1,這意味著每個 batch 包含一個材料結構。最後,初始學習率為 4×10-4,衰減速率為 0.5,衰減耐心為 20,最小選擇的學習率為 1×10-5,並在學習率達到此值時停止訓練。
DeepH 推理效能優異,可提供準確的能帶結構預測
在訓練、驗證和測試集上,模型預測的密度泛函理論哈密頓量矩陣元的平均絕對誤差 (MAE) 分別達到 1.45、2.35 和 2.20 meV, 這表明該模型具有對未曾見過的結構進行推理的能力。
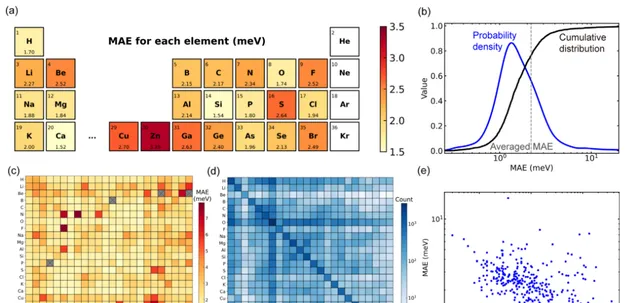
通用材料模型效能評估
在利用 104 種固體材料的大型材料資料庫對 Deep-2 方法訓練的通用材料模型進行效能評估時,在數據集的所有結構中,大約 80% 的材料結構具有小於平均值 (2.2 meV) 的平均絕對誤差。只有 34 個結構(約占測試集的 1.4%)的平均絕對誤差超過 10meV,說明該模型對主流結構有良好預測精度。
透過進一步分析數據集,模型在材料結構上的效能偏差可能是由於數據集分布偏差造成的。 研究發現,數據集中包含的元素對的訓練結構越多,相應的平均絕對誤差就越小。這一現象可能表明深度學習通用材料模型存在「縮放法則」,即更大的訓練數據集或許會提高模型效能。
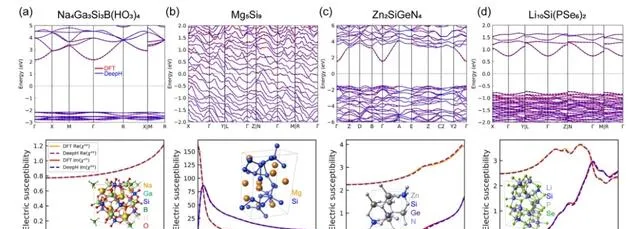
DFT 計算和 DeepH 預測的結果比較
為了評估 DeepH 通用材料模型預測材料性質的準確性,該研究在計算範例時,分別使用了基於密度泛函理論 (DFT) 計算和 DeepH 預測的 DFT 哈密頓量,然後將這兩種方法得到的計算結果進行了比較。結果表明, DeepH 預測的結果與 DFT 計算的結果非常接近,證明了 DeepH 在計算材料性質方面的出色預測精度。
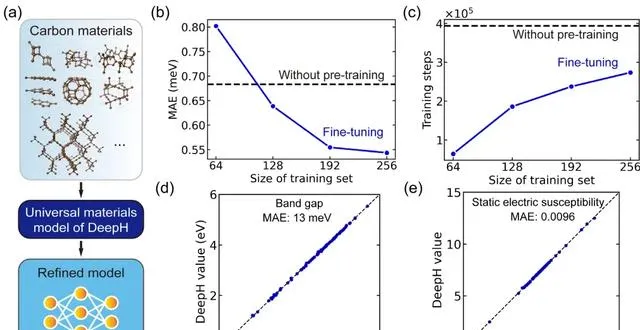
用於研究特定材料的微調通用材料模型
在具體的套用中,該研究用微調通用材料模型對碳同素異形體進行了研究。其中,碳材料數據集來源於薩馬拉碳同素異形體資料庫 (SACADA),共包含 427 種具有不同原子結構的碳同素異形體。
研究人員基於此對通用材料模型進行微調,建立了一個專門針對碳材料的改進型 DeepH 模型。與無預訓練模型相比,微調可以將預測 DFT 哈密頓量的平均絕對誤差顯著降低至 0.54 meV,還可以在少於 50% 的訓練結構中實作可比較的預測精度。
此外,微調還顯著改善了訓練收斂,並減少了訓練時間。可以說,微調有助於提高預測準確性並增強訓練效率。更重要的是, 微調後的 DeepH 模型在預測材料性質方面表現出了顯著的優勢,經過微調的模型幾乎可以為所有測試結構提供準確的能帶結構預測。
材料大模型風起雲湧, AI4S 任重道遠
以 ChatGPT 為時間起點,AI 正式進入了一個全新的「大模型時代」。這個時代的特點是利用龐大的數據集和先進的演算法,訓練出能夠處理復雜任務的深度學習模型。 在材料科學領域,這些大模型正與研究者們的智慧相結合,開啟了一個前所未有的研究新紀元。 這些大模型不僅能夠處理和分析海量的科學數據,還能夠預測材料的性質和行為,從而加速新材料的發現和開發,推動著這一領域向更高效、更精準的方向發展。
在過去的一段時間,AI for Science 正在與材料科學不斷碰撞出新的火花。
立足國內 ,北京凝聚態物理國家研究中心 SF10 組、中科院物理研究所、中科院電腦網路資訊中心共同合作,將數萬個化學合成路徑數據投餵給大模型 LLAMA2-7b,從而獲得了 MatChat 模型,可用來預測無機材料的合成路徑;電子科技大學聯合復旦大學、中國科學院寧波材料技術與工程研究所,成功開發出「耐疲勞鐵電材料」,在全球範圍內率先攻克困擾領域內 70 多年的鐵電材料疲勞問題;上海交通大學 AIMS-Lab 實驗室開發出了新一代材料智慧設計模型 Alpha Mat.……研究成果頻出,材料創新和發現進入新時代。
放眼全球 ,Google 旗下的 DeepMind 開發了用於材料科學的人工智慧強化學習模型 GNoME,尋找到了 38 萬余個熱力學穩定的晶體材料,相當於「為人類增加了 800 年的智力積累」,極大加快了發現新材料的研究速度;微軟釋出的材料科學領域人工智慧生成模型 MatterGen,可根據所需要的材料性質按需預測新材料結構;Meta AI 與美國高校合作,開發了行業頂級的催化材料數據集 Open Catalyst Project,以及有機金屬框架吸附數據集 OpenDAC……科技巨頭憑借自家的技術,將材料科學領域攪動的風起雲湧。
雖然與傳統材料研發方式相比,人工智慧為探索更廣泛的材料可能性開啟了大門,顯著減少了與材料發現相關的時間與費用。但是,AI for Science 在材料領域還面臨著可信度和有效實施的挑戰,確保數據品質、辨識和減輕用於訓練 AI 系統的數據潛在偏差等一系列問題有待解決。這或許也意味著,要想讓人工智慧在材料科學領域發揮更大的作用,仍然前路漫漫。











