2024年10月3日,J. Am. Chem. Soc.線上發表了威斯康辛大學麥迪遜分校Daniel C. Fredrickson 課題組的研究論文,題目為【 Machine Learning-Based Investigation of Atomic Packing Effects: Chemical Pressures at the Extremes of Intermetallic Complexity 】。

金屬間相代表了一個新興 行為的領域,其中具有堆積和電子偏好的原子可以組合成復雜的幾何排列,其長程 序涉及包含數千個原子的重復條紋,或者與三維晶胞不 相容 。 這種 排列 的形成表明了這些體系中無法解釋的驅動力, 如果理解 這些驅動力,就可以在新金屬材料的設計中加以利用 。 DFT化學壓力(CP)分析已成為 一種視覺化的方法,可以看到簡單晶體結構中的原子 堆積 張力如何驅動這種復雜性並創造潛在的功能 。 然而,由於該方法依賴於資源密集型的電子結構計算,其套用範圍迄今為止受到限制。
在此研究中,作者 利用金屬間反應性資料庫中的DFT-CP方案集開發了基於機器學習(ML)的CP方法實作 。透過一系列 例子 比較ML-CP和DFT-CP方案來說明該方法,然後透過探索金屬間化學中復雜的典型例項之一 Mg 2 Al 3 來演示其套用 ,Mg 2 Al 3 的高溫晶胞是一個包含1227個原子的2.8 nm立方體。對其ML-CP 衍生 的原子間壓力分析將結構的起源追溯到Frank-Kasper多面體組裝的簡單匹配規則。 ML-CP模型可以透過其 網路 界面或命令列版本立即用於其他金屬間化合物體系,只需一個晶體學資訊檔 。
所提出的ML-CP方法為將CP概念套用於新體系提供了一個框架,而不需要進行電子結構計算。在這個框架內,未來有大量發展的機會。 目前 機器學習模型的基礎僅在430種化合物和115種結構型別的2110個數據點上進行了訓練,沒有氫化物、鑭系元素和錒系元素的代表 。沿著這些思路,即使ML-CP模型的特征旨在允許對三元和四元體系進行預測,但訓練集中還沒有包含此類化合物(這意味著任何此類預測都應該進行批判性評估)。
雖然這個數據集足以為采樣的化合物領域生成一個訓練有素的模型,但金屬間反應性資料庫的擴充套件將允許對模型進行改進,以結合更廣泛材料的經驗。 此外,訓練中包含的大多數結構都是從文獻中提取的,代表了實驗觀察到的幾何形狀。 因此,目前尚不清楚該模型對異常或不穩定結構的預測有多準確,正如Cu 3 Ti擬合不佳所暗示的那樣。 期待 訓練ML-CP模型的未來叠代 以提高其通用性,同時探索其在原始DFT實作計算可行性之外的問題中的套用,如缺陷、晶界、非公度調變和準晶。
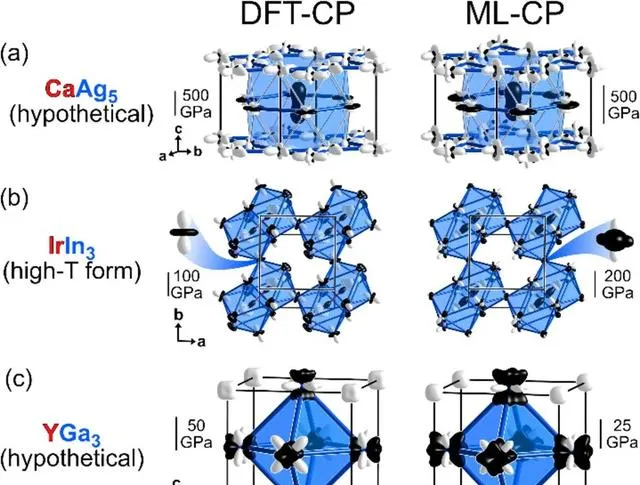
圖1 DFT-CP和ML-CP方案用於假設的CaCu 5 型CaAg 5 相、高溫CoGa 3 型IrIn 3 和AuCu 3 型YGa 3
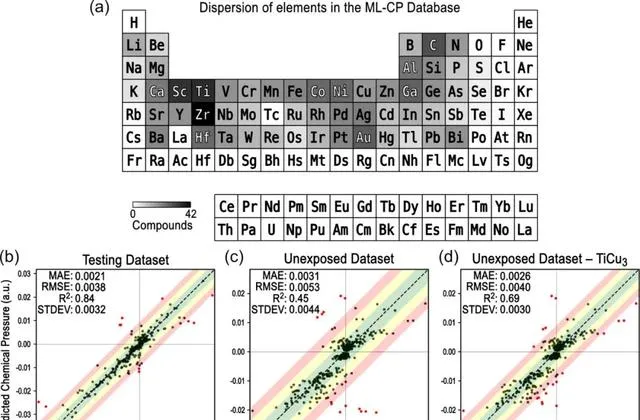
圖2 ML-CP模型的測試

圖3 Mg 2 Al 3 的低T菱方結構
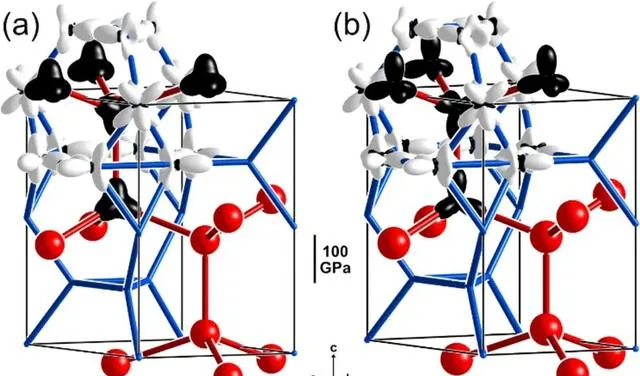
圖4 Mg-Al體系ML-CP模型的試驗

圖5 Mg 2 Al 3 中CP起伏的幾何起源
【 論文連結 】
Van Buskirk, J.S., Peterson, G.G.C. & Fredrickson, D.C . Machine Learning-Based Investigation of Atomic Packing Effects: Chemical Pressures at the Extremes of Intermetallic Complexity . J. Am. Chem. Soc. , 2024 . https://doi.org/10.1021/jacs.4c10479
【其他相關文獻】
[1] Sanders, K.M., Van Buskirk, J.S., Hilleke, K.P. et al. Self-Consistent Chemical Pressure Analysis: Resolving Atomic Packing Effects through the Iterative Partitioning of Space and Energy. J. Chem. Theory Comput ., 2023 , 19, 4273–4285. https://doi.org/10.1021/acs.jctc.3c00368
【註】:小編水平有限,若有誤,請聯系修改;若侵權,請聯系刪除!











